Se presenta caso de un paciente masculino de 16 años de edad con antecedentes de hematuria desde la infancia, además de alteraciones auditivas y oftalmológicas. Con esas características se sospecha síndrome de Alport, por lo que se inicia algoritmo diagnóstico, confirmando un síndrome de Alport con herencia autosómica recesiva debido a una homocigosis para la variación c.1131delC del gen COL4A3.
El objetivo de presentar este caso es revisar y resaltar las características de una enfermedad poco frecuente, considerarla dentro de los diagnósticos diferenciales en los pacientes que cursan con hematuria e insuficiencia renal de inicio temprano, resaltar la importancia de realizar un panel genético en lugar de estudiar gen por gen, y ser fuente de referencia para futuras investigaciones.
Palabras clave: Alport, gen col4a3, homocigosis.
ABSTRACT
The following is a case of a 16-year-old male patient with a history of hematuria since childhood, as well as hearing and ophthalmologic alterations. With these characteristics, Alport syndrome is suspected, is why diagnostic algorithm was started, confirming an Alport syndrome with autosomal recessive inheritance due to a homozygosis for the c.1131delC variation of the COL4A3 gene.
The objective of presenting this case is to review and highlight the characteristics of a rare disease, consider it within differential diagnoses in patients with hematuria and early renal failure, the importance of performing a genetic panel instead of studying gene by gene, and be a reference source for future research.
Keywords:Alport, gen col4a3, homozygosis
Introducción
El síndrome de Alport fue descrito por primera vez por Cecil Alport en 1927 como una “nefritis hemorrágica hereditaria familiar congénita” 1. Se trata de una enfermedad hereditaria de las membranas basales causada por mutaciones en la cadena de colágena tipo IV. Para establecer el diagnóstico se requiere muestras de tejido renal que muestran lamelación de la membrana basal glomerular, pero el Gold Standard diagnóstico son las pruebas genéticas 2. Clínicamente, los pacientes con síndrome de Alport se caracterizan por presentar nefropatía hereditaria progresiva comúnmente asociada a sordera sensorial, lesiones oculares y en ocasiones leiomiomatosis 3
El 80% de los pacientes tienen la forma ligada al cromosoma X, causada por mutaciones en el gen COL4A5 que codifica la síntesis de la cadena 5α del colágeno tipo IV 4. El 15% sufre la forma autosómica recesiva que surge de mutaciones en ambos alelos de los genes COL4A3 o COL4A4, responsables de la síntesis de las cadenas 3α (iv) y 4α (IV). El 5% restante padece la forma autosómica dominante debida a mutaciones heterocigotas en los genes COL4A3 o COL4A4, esta alteración también coincide con la encontrada en pacientes que sufren enfermedad de la membrana basal delgada 5Figura 1
El síndrome de Alport se caracteriza por hematuria, proteinuria significativa, hipertensión, sordera neurosensorial y progresión hacia insuficiencia renal crónica terminal. Estos síntomas son generales para todas las formas del síndrome de Alport; sin embargo, en el caso del síndrome ligado al X, los varones son los únicos en desarrollar la enfermedad mientras que las mujeres son portadoras, en ambos casos el diagnóstico es imprescindible. El síndrome de Alport con herencia autosómica recesiva se caracteriza por los síntomas clásicos y ocurre sin diferenciar sexo, debe sospecharse cuando un individuo exhibe el cuadro clínico y patológico de la enfermedad pero carece de antecedentes familiares, También debe sospecharse en casos de consanguinidad o personas procedente de poblaciones reducidas o aisladas. Las formas recesivas tienen un fenotipo severo, similar a la ligada al cromosoma X, mientras que las dominantes son más leves 6
Este síndrome se ha reportado en todos los grupos étnicos. Es la causa de Enfermedad renal crónica avanzada en 0,6 a 4,6% de los pacientes en Estados Unidos de Norteamérica y Europa. En Suecia, la frecuencia de hombres con la forma ligada al cromosoma x es de 1 en cada 17 000 nacidos vivos. No existe casuística al respecto en Ecuador 3
REPORTE DE CASO
Paciente masculino de 16 años de edad acude a la consulta externa, en noviembre de 2014, referido unidad de atención de primer nivel por elevación de los niveles de azoados (urea: 63,9 mg/dl, creatinina: 2,24 mg/dl), proteinuria 24 h: 1346,10 mg/24 horas, así como disminución de la agudeza visual y auditiva.
Al ingreso, el paciente dispone de los resultados de una biopsia renal realizada en la unidad hospitalaria de referencia que reporta Nefritis túbulo-intersticial difusa, nefroangioesclerosis mayor del 50%, atrofia tubular, glomerulonefritis membranosa con semilunas fibrosas menor de 50% y hialinosis mayor de 25%.
El paciente presento enfermedad renal crónica en estado III A 3, por lo que recibió tratamiento antiproteinurico con espironolactona 50 mg y enalapril 10 mg.
Se realizó audiometría, confirmando la disminución de la capacidad auditiva del paciente, y diagnosticando sordera neurosensorial. Por otro lado, en exámenes complementarios se evidencio catarata cortical anterior que ocluye parcialmente el eje visual del ojo derecho, mientras que el ojo izquierdo se presenta sin alteraciones. El fondo de ojo también fue normal en ambos ojos.
Ante la sospecha de síndrome de Alport se le realizó el estudio genético, en busca de mutación del gen COL4A5, que es la presentación más frecuente. Y el estudio de la secuenciación de dicho gen resulto negativo, por lo que se estudiaron otros genes implicados al colágeno tipo IV, COL4A3, COL4A4, COL4A5 y COL4A6, detecto una mutación en homocigosis en el gen COL4A3. Esta mutación consistió en una deleción de una base nitrogenada (citosina) en la posición 1131 del gen, lo cual provoca un cambio del marco de lectura y por tanto una alteración en la proteína correspondiente a p.Gly378Glufs*22. Además se realizó estudio genético familiar y asesoría genética a los familiares, resultando dos varones afectos de un total de tres hermanos. Figura 2
EVOLUCIÓN
En marzo de 2016, el paciente progresa a Enfermedad Renal Crónica avanzada, y 7 meses más tarde el paciente ingresa al programa de diálisis peritoneal. Fue evaluado para trasplante renal, el mismo que se realizó 8 meses luego de haber ingresado a la lista de espera única nacional, su evolución fue favorable con creatinina actual de 1,22mg/dL.
DISCUSIÓN
El caso descrito clínicamente presenta todas las características del síndrome de Alport que incluyen alteraciones de la función renal, auditivas y oftalmológicas 2. Sin embargo, debe tomarse en cuenta que el paciente no tiene antecedentes familiares de pérdida de la función renal.
En las clasificaciones actuales de la enfermedad, el síndrome de Alport con herencia autosómica recesiva se ha asociado a mutaciones en los genes COL4A3 o COL4A4 e implica un 100% de riesgo de Enfermedad Renal Crónica Avanzada, con una tasa de progresión y tiempo de manifestaciones extra renales influenciado por el genotipo 7
El procedimiento diagnóstico habitual son las pruebas genéticas, que tienen un 90% de sensibilidad 2, son el Gold Standard y una herramienta imprescindible para dar el diagnostico. En la actualidad las pruebas genéticas generalmente reemplazan las investigaciones más invasivas, como la biopsia renal, y es probable que logren la confirmación del diagnóstico a nivel molecular de los pacientes con Síndrome de Alport 8
La mayoría de los laboratorios utilizan la secuenciación de última generación por paneles genéticos para detectar simultáneamente los genes correspondientes a las cadenas α3, 4 y 5 del colágeno tipo IV. Siendo el Gold Standard no solo por la velocidad del análisis, sino también porque asegura la detección de las formas digénicas, es decir, aquellas formas debidas a mutaciones en dos genes de colágeno de tipo IV diferentes. Este paciente ilustra la pertinencia de este abordaje genético ya que se consideró inicialmente la forma recesiva ligada al X del síndrome de Alport en virtud de su mayor frecuencia poblacional y de la coexistencia de dos varones afectos en esta familia. Sin embargo, el resultado negativo de este estudio sugirió la necesidad de ampliar el estudio genético a un panel de otros genes también asociados con este síndrome, ocasionando de esta manera, definición diagnóstica en mayor tiempo. Si la secuenciación es negativa, los médicos pueden recurrir a la biopsia renal (con microscopía electrónica e inmunofluorescencia), biopsia de piel, análisis de ARN (obtenidos de la piel o la raíz del pelo) o pruebas genéticas adicionales 8
En los pacientes con síndrome de Alport se debe valorar indicadores de mal pronóstico como hipertensión, proteinuria, insuficiencia renal, e iniciar tratamiento. Incluir un inhibidor de enzima convertidora de angiotensina (IECA) puede retrasar la evolución de la enfermedad a etapa terminal y mejorar la esperanza de vida 9. En este caso, a pesar del inicio temprano del tratamiento con IECA, la progresión de la enfermedad fue inevitable.
Las personas con esta enfermedad tienen una supervivencia del injerto mejor que la población general con enfermedad renal en etapa terminal. Sin embargo, estos pacientes están en riesgo de enfermedad anti-GBM post-trasplante debido a la formación de autoanticuerpos, hallazgo relevante a tener en cuenta durante el seguimiento de los pacientes8. A pesar de esto, el trasplante no debe contraindicarse ya que ofrece una mejor calidad de vida que la diálisis en gran parte de la población joven 10
Con esta notificación se reporta primer caso síndrome de Alport con herencia autosómica recesiva debido a una homocigosis para la variación c.1131delC del gen COL4A3 en Ecuador, resaltado la importancia de la asesoría genética y reproductiva que va implícita a un diagnóstico de síndrome de Alport.
AGRADECIMIENTOS
Agradecemos profundamente a todo el personal que labora en el departamento de Nefrología, y al departamento de Genética del hospital.
CONFLICTO DE INTERESES.
Los autores declaran que no tienen conflictos de intereses sobre el material aquí publicado.
Presentación Atípica de la Glomerulonefritis Membranoproliferativa Idiopática en Adulto - Relato de Caso y Revisión Bibliográfica
Autores:
Francisco Schossler Loss
Karina Litcheteneker
Fernando Roberto Roman
Sérgio Kazuyuki Saito
Resúmen
La glomerulonefritis membranoproliferativa (GNMP), conocida como glomerulonefritis mesangiocapilar, presenta en la biopsia renal hipercelularidad y espesamiento de la membrana basal glomerular formando doble contorno en las paredes de los capilares glomerulares y proliferación endocapilar, generalmente llevando la apariencia lobular del tufo glomerular en la microscopía electrónica. Describimos el caso de un paciente masculino, 29 años, obeso, sin enfermedades previas que inició súbitamente con cuadro de disuria, hematuria macroscópica y dolor de media intensidad en flanco derecho. En biopsia renal presentó en el anatomopatológico conformación de Glomerulonefritis Membranoproliferativa, pero en la inmunofluorescencia no presentó depósitos de inmunocomplejos.
Membranoproliferative glomerulonephritis (GNMP), known as mesangiocapillar glomerulonephritis, presents in the renal biopsy hypercellularity and thickening of the glomerular basement membrane forming a double contour on the walls of the glomerular capillaries and endocapillary proliferation, generally leading to the lobular appearance of the glomerular tuft in electron microscopy¹. We describe the case of a 29-year-old obese male patient with no prior disease who started abruptly with dysuria, macroscopic hematuria and medium intensity pain in the right flank. In renal biopsy he presented in the anatomopathological conformation of Membranoproliferative Glomerulonephritis, but in immunofluorescence he did not present deposits of immunocomplexes.
El paciente, de 29 años, obeso - IMC 31,9, previamente hígido, inició abruptamente con disuria, hematuria macroscópica con dismorfismo eritrocitario y dolor de media intensidad en flanco derecho. En la mayoría de los casos, se observó una disminución de la función renal, creatinina 5,7 mg / dl y urea 162 mg / dl, piuria con cilindros leucocitarios, proteinuria (< 300 mg), anasarca e hipertensión arterial sistémica. En la mayoría de los casos, se observó un aumento en la concentración de la aorta (40 mm³), la fracción de eyección preservada, después de cinco días de internación, presentó cuadro de pleurite - dolor ventilatorio - dependiente y fricción pleural con pequeño derrame pericárdico evidenciado en el ecocardiograma (9 mm³) además de la dilatación de la raíz de la aorta (40mm³). Sorologías virales, factor antinuclear (FAN) y factor reumatoide negativo; otros exámenes para investigación de GNRP aún no habían sido liberados por el laboratorio. Por la historia clínica, se sospechó de Glomerulonefritis Rapida Progresiva (GNRP) y se inició pulsoterapia con corticoide - metilprednisolona 1 gramo por tres días - y mantenimiento con prednisona 60 mg / día con mejora de la función renal. Por la mejora del cuadro con estabilización de la función renal, el paciente recibió alta hospitalaria con prednisona 60 mg / día y furosemida 120 mg / día además de sintomáticos, siendo acompañado ambulatoriamente.
Después de quince días, el paciente retorna al ambulatorio con parcial de orina evidenciando sólo rasgos de proteínas y remisión de la hematuria post-corticoide. Además de los demás exámenes negativos: Anti-DNA, Anti-Smith, Anti-SSA; crioglobulinas y FAN no reactivos; C3 (163 mg / dl) y C4 (43 mg / dl) ambos normales. Con ello y por el buen estado general, se mantuvo prednisona 60mg / día, furosemida 80mg / día y omeprazol 40mg / día.
Después de 45 días de seguimiento ambulatorio, el paciente inició súbitamente con anasarca, hipotensión (90/60 mmHg), crisis convulsivas - tomografía computarizada evidenció lesiones isquémicas en lobo frontal sugestivas de vasculitis, plaquetopenia - 80.000 mm² y urticaria, siendo internado en la Unidad de Terapia Intensiva (UTI) e iniciada la investigación para Lupus Eritematoso Sistémico, el cual no fue confirmado. Figura 1 - imagen del abdomen del paciente durante el primer internamiento, figura 2 - después de la apertura del cuadro abrupto de urticaria y plaquetopenia. En los exámenes de laboratorio de esta segunda admisión: urea 2011mg / dl, creatinina 9,2 mg / dl, sodio y potasio normales, bilirrubina total 3,0 g / dl; fosfatasa alcalina, amilasa, lipasa, gamma glutamil transferasa, aspartato transaminasa y aspartato aminotransferasa, RNI, albúmina normal; deshidrogenasa láctica 501 U / L, proteína C- Reactiva 151mg / dl. Parcial de orina con hematuria dismórfica, piuria y proteinuria (< 300mg). Todos los demás exámenes estaban en el rango de la normalidad o no - reactivos: C3 y C4, citomegalovirus IgG e IgM, coombs indirecto, Epstein BARR, anti-DNA, Anti-HCV, FAN, P- ANCA, C- ANCA, toxoplasmosis IgG, urocultura, VDRL y sedimentación. En este cuadro se inició dexametasona 4mg endovenoso (EV) 6/6 horas, fenitoína 100mg vía oral (VO) 8/8 horas y carbamazepina 200mg VO 8 / 8horas. El paciente se quedó 17 días internado en la UTI, mejoró del cuadro y fue encaminado a biopsia renal.
En el anatomopatológico de la biopsia renal - figuras 3, 4 y 5 - se evidenció cortes del tejido renal cortical conteniendo hasta 13 glomérulos con celularidad mesangial discretamente aumentada y acompañada de expansión de la matriz mesangial, con túbulos tróficos y yuxtapuestos. Interstício delicado y sin infiltrados inflamatorios, ramas arteriolares sin alteraciones histológicas. Siendo así, el paciente fue diagnosticado con Glomerulonefritis Membranoproliferativa con Expansión de la Matriz Mesangial. En la secuencia, se analizó la biopsia renal de la lámina de la inmunofluorescencia directa que relató ausencia de depósitos de inmunoglobulinas y complemento, siendo que los hallazgos de inmunofluorescencia no permitieron caracterizar lesión mediada por inmunocomplejos o anticuerpos antimembrana basal en actividad. Se han investigado IgG, IgM, IgA, C3, C1q, fibrinógeno, Kappa y Lambda.
Después de un año del inicio de los síntomas, el paciente sigue en seguimiento ambulatorio mensualmente. Está en uso de micofenolato mofetilo 500mg 2 cápsulas 12 / 12h, atorvastatina 20mg / día, meticorten 20mg 1 comprimido y medio al día, carbamazepina 200mg 12 / 12h, carbonato de calcio 500 mg 2 comprimidos al día, vitamina D 7000ui VO en la semana y losartana 50mg + hidroclorotiazida 12,5mg. Actualmente el paciente está estable, con todos los exámenes de laboratorio dentro de la normalidad y no presentó nuevo agravamiento de la enfermedad.
Discusión
La glomerulonefritis membranoproliferativa (GNMP) se caracteriza por la proliferación mesangial, la expansión de la matriz y su interposición entre el endotelio y la membrana basal glomerular, proporcionando la apariencia de doble contorno. Se clasifica en tres tipos según la ubicación del depósito denso en electrones a la microscopía electrónica: tipo I (depósitos subendoteliales); tipo II (depósitos intramembranosos homogéneos densos); tipo III (variante del tipo I, con depósitos subepiteliales y subendoteliales). El tipo I fue asociado a la infección por el virus de la hepatitis C (VHC), mientras que el tipo II afecta a individuos más jóvenes y no estaba relacionado con causas sistémicas 1,4.
Esta glomerulopatía está frecuentemente asociada a causas secundarias, principalmente la infección por el VHC y enfermedades autoinmunes, tales como lupus eritematoso sistémico (LES) y artritis reumatoide. Sin embargo, se identificaron un 34% de formas idiopáticas en un grupo de pacientes predominantemente del sexo masculino, con edad de 24,9 años con síndrome nefrótico y bajos niveles de fracción de complemento C3 en la mitad de ellos 3,8.
Una nueva clasificación fue propuesta por Sethi et al 1, en el cual el sistema de clasificación de GNMP se basó en inmunofluorescencia (IF), definiendo dos grupos: GNMP con deposición de inmunoglobulina en la IF, que podría estar asociada a enfermedades autoinmunes, infecciones o gamopatía monoclonal; mientras que la GNMP sin deposición de inmunoglobulina, pero con deposición de C3 en la IF, que se clasifica como enfermedad de depósito denso (DDD) o glomerulonefritis C3 después del examen por microscopía electrónica 1,8. En otro estudio, la GNMP con ninguna deposición observada en la IF fue introducida en otro grupo, y puede ser secundaria a reacciones de membrana, como en la microangiopatía trombótica 5. En esta nueva clasificación, el uso de microscopía electrónica es obligatorio en casos de deposición exclusiva de C3, a fin de diferenciar la DDD y glomerulonefritis C3 1,8. Algunos autores creen que las formas idiopáticas deben ser muy raras, enfatizando el complemento y los estudios genéticos en esos pacientes para esclarecer sus diagnósticos 6. La GPMN idiopática es uno de los tipos menos comunes de glomerulonefritis, responsable de aproximadamente el 4 y el 7% de las principales causas renales del síndrome nefrótico en niños y adultos, respectivamente 9. Nuestro paciente se encuadra en la forma idiopática, demostrando la rareza del caso. La crioglobulinemia esencial mixta asociada a la infección por el VHC ha sido implicada en un número sustancial de casos de lo que se pensaba que era MPGN idiopático 7. Estos factores no se encontraron en nuestro paciente.
La glomerulonefritis membranoproliferativa afecta principalmente a niños y adultos jóvenes, sin predilección por sexo 8. En niños, la GNMP es a menudo idiopática, mientras que en adultos la GNMP se asocia comúnmente a la crioglobulinemia ya la infección por el VHC 8. Las manifestaciones clínicas más comunes en pacientes con GNMP son: síndrome nefrótico (40-70%); síndrome nefrótico agudo (20-30%); proteinuria asintomática y hematuria detectada en la parcial de orina de rutina (20-30%); episodios recurrentes de hematuria macroscópica (10 a 20%) 8.
La infección del tracto respiratorio puede preceder al diagnóstico en la mitad de los pacientes. La hipertensión se encuentra en la presentación en un tercio de los pacientes, sin embargo es más frecuente con enfermedad progresiva. La disfunción renal ocurre en más del 50% de los pacientes. En comparación con los adultos, los niños son más propensos a tener hematuria al principio y menos propensos a tener insuficiencia renal e hipertensión 8,11.
La hipocomplemia es la característica común de todos los tipos de GNMP, que es esencialmente la única causa de síndrome nefrótico idiopático que está asociado a la hipocomplemia. En GNMP tipo I y GNMP crioglobulinémica, la vía clásica es preferentemente activada (C3 bajo o normal, C4 bajo y CH 50), mientras que en la MPGN tipo II, la vía alternativa es activada (C3 bajo, C4 normal y CH 50 bajo), y en el tipo III GNMP, el perfil del complemento típicamente revela evidencia de la activación tanto de la vía alternativa y de la vía terminal (baja C3, normal C4 y baja C5 a C9) 12,13.
Por lo tanto, para evaluar a un paciente con sospechosos de GNMP debemos atentar a los hallazgos históricos, como: infección del tracto respiratorio superior, síntomas urinarios (oliguria, hematuria), síntomas de anemia (fatiga, palidez), síntomas urémicos (anorexia, vómito) , síntomas sugestivos de GNMP secundaria (ictericia, dolor articular, pérdida de peso). En el examen físico evaluar la presión sanguínea, signos de síndrome nefrótico, signos de enfermedad hepática crónica, neuropatía periférica. En los exámenes de laboratorio evaluar creatinina, proteína de 24h, electroforesis de inmunoglobulina, colesterol sérico, lactato, hemograma completo, complementos (C3, C4, CH50), ANCA, factor reumatoide, crioglobulinas, virus de la hepatitis C, virus de la hepatitis B, virus de la hepatitis B, virus de la hepatitis B, inmunodeficiencia humana y hemocultivos. Examen de imagen como ultrasonografía renal y, por último, el histopatológico con biopsia renal 8.
No existe tratamiento estándar para la GNMP, pues los mayores ensayos realizados hasta la fecha fueron descontrolados e incluyeron pacientes de los tres tipos de GNMP en diferentes proporciones, dificultando el análisis de los estudios 14. Planes terapéuticos para glomerulonefritis membranoproliferativa idiopática son controvertidos e incluyeron corticosteroides, inmunosupresores , antiplaquetarios y agentes biológicos 8.
El corticoide demostró tener efecto benéfico en días alternos en la sobrevida renal en pacientes pediátricos con GNMP 17. Sin embargo, su uso prolongado puede tener efecto adverso, como alteración del crecimiento óseo 18. En ese sentido, la corticoterapia se mostró eficaz en el tratamiento de niños, sin embargo no se encontraron evidencias convincentes que esteroides sean eficaces en modificar la actividad de la enfermedad o la progresión de la enfermedad en adultos con GNMP idiopática 8.
El micofenolato mofetilo (MMF) es un agente antiproliferativo utilizado en el tratamiento de la enfermedad renal mediada inmunológicamente 8. En la GNMP idiopática, estudios preliminares sugieren que la combinación de MMF y corticoesteroides puede reducir a corto plazo la proteinuria y preservar la función renal 15, 16.
La ciclofosfamida es eficaz en la remisión y en la interrupción de la progresión de la GNMP a la enfermedad renal crónica terminal, principalmente si está asociada a pulso de metilprednisolona y prednisolona oral. Se recomienda, también, la ciclofosfamida en pacientes con glomerulonefritis rápidamente progresiva o con deterioro reciente de la función renal 19.
Por el consumo aumentado de plaquetas y por la posible lesión glomerular generada por ellas en la GNMP, se indica la terapia antiplaquetaria, mejorando la función renal y la tasa de proteinuria 20,21.
Consideraciones finales
Debemos evaluar cuidadosamente la clínica del paciente y no sólo los exámenes que se evidencian. En este caso, el paciente abrió el cuadro con una glomerulonefritis rápidamente progresiva, siendo controlada sólo con pulsoterapia de corticoide y diurético. Sin embargo, la causa base de su patología estaba en la Glomerulonefritis Membranoproliferativa Idiopática, evidenciada en la microscopia. El paciente tuvo la estabilización del cuadro por completo y la mejora total de los exámenes de laboratorio que estaban alterados después de la asociación de corticoide con micofenolato de mofetilo, siendo así, en esos casos, indicamos que se inicie lo más pronto posible la asociación de estos medicamentos.
NEFROPATÍA POR IgA EN EL LUPUS ERITEMATOSO SISTÉMICO: REPORTE DE UN CASO.
IgA NEPHROPATHY IN SYSTEMIC LUPUS ERYTHEMATOSUS: A CASE REPORT
Autores:
Dra. Teresa Moreira
Dr. Vasco Batista
Dr. Carlos Botelho
Correspondencia:
Dra. Teresa Moreira Internal Medicine Department, Centro Hospitalar do Tâmega e Sousa, Penafiel, Portugal. teresa_margaridam@hotmail.com Avenida do Hospital Padre Américo 210, 4564-007 Guilhufe 00351915575343
Resumen:
La afectación renal en el lupus eritematoso sistémico es uno de los aspectos más típicos de la enfermedad. Raramente se ha notificado nefritis no lúpica en estos pacientes. Aunque la nefropatía por IgA y la nefritis por lupus comparten algunas características fisiopatológicas comunes, su laboratorio, histopatología y hallazgos clínicos extrarrenales son diferentes y apoyan una patogénesis diferente.
Presentamos el caso de una paciente con un diagnóstico clínico bien establecido de lupus eritematoso sistémico en el que la biopsia renal reveló inesperadamente nefropatía por IgA, sin características superpuestas de nefritis por lupus.
Renal involvement in systemic lupus erythematosus is one of the most typical aspects of the disease. Non-lupus nephritis has rarely been reported in these patients. Although IgA nephropathy and lupus nephritis share some common physiopathological characteristics, their laboratory, histopathology and extrarenal clinical findings are different, and support a different pathogenesis.
We present the case of a female patient with a well established clinical diagnosis of systemic lupus erythematosus in whom the renal biopsy unexpectedly revealed IgA nephropathy, without any superimposed features of lupus nephritis.
Key words: Anti -Ro/SSA antibodies, autoimmunity, IgA nephropathy, lupus nephritis, renal biopsy, systemic, lupus erythematosus.
Introducción
La afectación renal en el lupus eritematoso sistémico (LES) es uno de los aspectos más típicos de la enfermedad. Las deposiciones inmunes se encuentran en casi todas las biopsias renales de pacientes con LES, reflejando la alta prevalencia de compromiso renal, incluso en ausencia de manifestaciones clínicas evidentes 1.
La morfología de la nefritis lúpica abarca un amplio espectro en diferentes pacientes, así como en biopsias seriadas del mismo paciente e incluso entre diferentes glomérulos de la misma biopsia 1,2.
La nefropatía lúpica (NL) se clasifica según las alteraciones glomerulares en microscopía óptica integrada con patrones de inmunofluorescencia y microscopía electrónica 3. Las muestras de biopsia en serie muestran que la transformación de la clase de histología es frecuente 4. Por lo general, la NL presenta inmunoglobulina policlonal, distribución de los factores del complemento C1q, C3 y C4, depósitos de densidades inmunológicas como "huella dactilar", cuerpos de hematoxilina, lesiones en “asas de alambre" y depósitos inmunitarios frecuentes identificados en la membrana basal capilar 2-4.
La nefropatía por IgA (IgAN) es la glomerulonefritis primaria más frecuente, caracterizada por la proliferación mesangial y los depósitos mesangiales difusos de IgA 5.
Desde la primera descripción de Berger et al. (1967), la nefropatía por IgA se ha encontrado típicamente en asociación con enfermedades hepáticas y púrpura de Schönlein-Henoch, pero en los últimos años se han reportado otras asociaciones, principalmente con trastornos sistémicos, como las enfermedades reumáticas (espondilitis anquilosante, artritis reumatoide, síndrome de Reiter, uveítis, síndrome de Behçet, síndrome sicca), crioglobulinemia y otras enfermedades relacionadas con la piel y el tracto respiratorio superior y gastrointestinal, entre otros órganos 5. Algunas de estas asociaciones se han reportado solo en casos aislados, sugiriendo que pueden ser puramente coincidentes.
Varias enfermedades pueden ser acompañadas de una desregulación del sistema inmunitario IgA, lo que implica la presencia de IgA polimérica sérica aumentada o de complejos inmunitarios IgA circulantes, y la consiguiente deposición en el riñón 5. La superposición y la aparición de otras glomerulopatías no lúpicas es rara en pacientes con LES.
La biopsia renal desempeña un papel crucial en la identificación de estas glomerulopatías, que pueden tener implicaciones pronósticas y terapéuticas distintas de las de la nefritis lúpica.
INTRODUCTION
Renal involvement in systemic lupus erythematosus (SLE) is one of the most typical aspects of the disease. Immune depositions are found in nearly all renal biopsies of SLE patients, reflecting the high prevalence of renal involvement, even in the absence of overt clinical manifestations 1.
The morphology of lupus nephritis spans a wide spectrum in different patients as well as in serial biopsies of the same patient and even among different glomeruli of the same biopsy 1,2.
iLupus nephropathy (LN) is classified based on glomerular alterations on light microscopy integrated with immunofluorescence and electron microscopy patterns 3. Serial biopsy specimens show that transformation of histology class is frequent 4. Typically, LN presents polyclonal immunoglobulin, distribution of complement factors C1q, C3 and C4 with “fingerprint” immune dense deposits, haematoxylin bodies, “wire-loop” lesions and frequent immune deposits identified in the capillary basement membrane 2-4.
IgA nephropathy (IgAN) is the most common primary glomerulonephritis, characterized by mesangial proliferation and diffuse mesangial IgA deposits 5.
Since the first descript ion of Berger et al. (1967), IgA nephropathy has been found typically in association with liver diseases and Henoch-Schönlein purpura, but in the past few years other associations have been reported, mainly with systemic disorders, such as rheumatic diseases (ankylosing spondylitis, rheumatoid arthritis, Reiter syndrome, uveitis, Behcet’s syndrome, sicca syndrome), cryoglobulinemia, and with other illness involving skin and upper respiratory and gastrointestinal tract, among other organs 5. Some of these associations have been reported only in isolated cases, suggesting that they may be purely coincidental.
Several diseases may be accompanied by a dysregulation of the IgA immune system, implying the presence of increased serum polymeric IgA or of circulating IgA immune complexes, and consequent deposition within the kidney 5. The superimposition and occurrence of other nonlupus glomerulopathies is rare in SLE patients.
Renal biopsy plays a crucial role in identifying these glomerulopathies, which may have prognostic and therapeutic implications distinctive from those of lupus nephritis.
Reporte de Caso
Una mujer caucásica de 24 años, no fumadora, sin enfermedades graves previas, fue referenciada a nuestra clínica en octubre de 2015 con un historial reciente (un mes) de aumento de la debilidad muscular, fiebre, fotosensibilidad y poliartritis simétrica en las articulaciones pequeñas de las manos y muñecas. El examen clínico reveló úlceras orales y una erupción cutánea en la región malar que había aparecido en los 3 meses anteriores, sin otros hallazgos significativos. Los análisis de sangre de laboratorio revelaron un aumento de la tasa de sedimentación de eritrocitos (TSH 72mm/h) y leucopenia (recuento de glóbulos blancos 3.20 × 103 / μL). El análisis de orina fue negativo para proteínas y sangre. Los cultivos de sangre y orina fueron negativos. Los estudios inmunológicos revelaron anticuerpos anti-Ro / SSA fuertemente positivos, con resultados negativos para anticuerpos antinucleares (ANA), anti-dsDNA, anti-Sm, anti-RNP y antifosfolípidos y ANCA. Los niveles séricos de complemento C3 y C4 disminuyeron ambos. El factor reumatoide y las pruebas de laboratorio para la sífilis y el antígeno de superficie de la hepatitis B (HBsAg) fueron negativos. Los hallazgos clínicos y bioquímicos cumplieron con los criterios diagnósticos del LES del American College of Rheumatology y, en conjunto, permitieron su diagnóstico. En ese momento no había evidencia de enfermedad renal. Se inició tratamiento con prednisolona (10 mg por día) e hidroxicloroquina (400 mg por día), con mejoría posterior de la artritis y signos cutáneos, y normalización de los análisis de sangre de laboratorio (TSH, recuento de glóbulos blancos, C3 y C4 séricos).
Las pruebas de función renal y el análisis de orina se mantuvieron normales. En noviembre de 2016, un análisis de orina de rutina mostró proteinuria y hematuria microscópica. La microscopía de orina mostró 10 eritrocitos y 2-4 moldes granulares eritrocitos por campo de gran aumento. La excreción de proteínas en orina de 24 horas fue de 1.2 g. Se realizó una biopsia renal percutánea con aguja. El examen histológico mostró esclerosis en 6 de los 9 glomérulos, proliferación mesangial moderada, atrofia tubular y fibrosis intersticial difusa Figura 1. La tinción de inmunofluorescencia de 6 glomérulos reveló depósitos mesangiales de IgA y C3 con ausencia de IgG, IgM, C1q, C4 y fibrinógeno Figura 2. Se estableció el diagnóstico de IgAN.
Se mantuvo el tratamiento previo y se inició el tratamiento con ramipril 5 mg por día, con una disminución de la excreción de proteínas en orina de 24 horas a 0,8 g y desaparición de la hematuria 3 meses después de la biopsia renal. La función renal se mantuvo normal. En ese momento, se decidió agregar losartán 25 mg por día.
Actualmente la paciente permanece asintomática, con presión arterial normal. La función renal es normal y la excreción de proteínas en la orina durante 24 horas ha disminuido a 0,3 g. El recuento de glóbulos blancos, la TSH y las fracciones C3 y C4 del complemento sérico son normales, los títulos de ANA son indetectables, los niveles séricos de IgG, IgA e IgM son normales y los anticuerpos anti-Ro / SSA siguen siendo positivos.
CASE REPORT
A 24-year-old, non-smoking caucasian woman, with no previous serious illnesses, was referred to our clinic in October 2015 with a recent (one month) history of increased muscle weakness, fever, photosensitivity and symmetric polyarthritis involving the small joints of the hands and wrists. Clinical examination revealed oral ulcers and a skin rash on the malar region that had appeared in the previous 3 months, with no other significant findings. Laboratory blood tests revealed an increase of the erythrocyte sedimentation rate (ESR 72 mm/1sth) and leukopenia (white cell count 3.20×103/μL). The urine analysis was negative for protein and blood. Blood and urine cultures were negative. Immunological studies revealed strongly positive Anti -Ro / SSA antibodies, with negative results for antinuclear (ANA), ds DNA, anti-Sm, anti -RNP and antiphospholipid antibodies and ANCA. Serum complement C3 and C4 levels were both decreased. Rheumatoid factor and laboratory tests for syphilis and hepatitis B surface antigen (HBsAg) were negative. The clinical and biochemical findings met the American College of Rheumatology diagnostic criteria for SLE, and taken together allowed its diagnosis. At that time there was no evidence of renal disease. Treatment with prednisolone (10 mg per day) and hydroxychloroquine (400 mg per day) was started, with subsequent improvement of the arthritis and cutaneous signs, and normalization of laboratory blood tests (ESR, white cells count, serum C3 and C4).
Renal function tests and urine analysis remained normal. In November 2016, a routine urine analysis showed proteinuria and microscopic hematuria. Urine microscopy showed 10 erythrocytes and 2-4 granular casts per high -powerfield. 24 -hour urine protein excretion was 1.2 g. A percutaneous needle kidney biopsy was performed. Histological examination at light showed sclerosis in 6 of the 9 glomeruli, modest mesangial proliferation, tubular atrophy and diffuse interstitial fibrosis (Figure 1). Immunofluorescence staining of 6 glomeruli revealed mesangial deposits of IgA and C3 with absence of IgG, IgM, C1q, C4 and fibrinogen (Figure 2). The diagnosis of IgAN was established.
Previous therapy was maintained and treatment with ramipril 5 mg per day was started, with a decrease in 24 -hour urine protein excretion to 0.8 g and disappearance of hematuria 3 months after kidney biopsy. Renal function remained normal. At that time, it was decided to add losartan 25 mg per day.
Presently the patient remains asymptomatic, with normal blood pressure. Renal function is normal and 24-hour urine protein excretion has decreased to 0.3 g. White cell count, ESR and serum complement C3 and C4 fractions are normal, ANA titers are undetectable, serum IgG, IgA and IgM levels are normal and anti -Ro/SSA antibodies remain positive.
DISCUSION
IgAN se ha descrito anteriormente en asociación con varias enfermedades autoinmunes, pero según nuestro conocimiento, se han notificado pocos casos previos de IgAN asociados con LES 1,8,9. A pesar de ser enfermedades distintas, con diferentes hallazgos clínicos y patológicos y con diferentes pronósticos, comparten algunas características fisiopatológicas comunes 1,8,9.
La afectación renal en el LES se caracteriza por grados variables de proteinuria, cambios en los sedimentos urinarios con microhematuria y cilindros de eritrocitos, hipertensión y disfunción renal progresiva.
Los signos extraorales típicos en la piel, las articulaciones y la sangre a menudo se asocian con la NL, que generalmente permite una fácil diferenciación de la IgAN. Sin embargo, en IgAN también puede haber algunas características extra-renales, como artralgia, lesiones vasculíticas de la piel y eritema que confunde con LES 9,10.
La IgAN es la causa más común de glomerulonefritis idiopática en el mundo 11. Aunque inicialmente se pensó que este trastorno tenía un curso benigno, ahora se reconoce que la progresión lenta hacia la etapa final de la enfermedad renal ocurre en hasta el 50 por ciento de los pacientes afectados 11. La patogenia de IgAN es incierta y su etiología es desconocida. La regulación inmune anormal da como resultado la formación de complejos inmunes que contienen IgA caracterizados por su alta afinidad por el mesangio 5.
LES es claramente una enfermedad autoinmune; los intentos de relacionar la IgAN con la autoinmunidad han fracasado debido a la falta de autoanticuerpos significativos en la IgAN 12.
Los aspectos inmunohistológicos de IgAN se caracterizan por la proliferación mesangial y los depósitos mesangiales de IgA1 (generalmente ausentes en la nefritis lúpica), la fracción de complemento C3 y, en ocasiones, la IgG y la IgM, que son responsables de la activación del complemento con la consiguiente liberación de mediadores inflamatorios 5. Aunque el cuadro clínico puede ser muy sugerente, el diagnóstico solo puede confirmarse mediante una biopsia de riñón.
Nuestra paciente tenía características clínicas características de LES, con cinco criterios diagnósticos del American College of Rheumatology. Según nuestro conocimiento, nuestra paciente no tenía antecedentes familiares que sugirieran una afección hereditaria como la inmunodeficiencia.
Dos años después del diagnóstico clínico de LES, se detectó afectación renal (proteinuria leve y hematuria microscópica con una función renal conservada) y el paciente se sometió a una biopsia renal. En nuestro caso, la biopsia renal no mostró hallazgos histológicos típicos de LES.
Se puede argumentar que el predominio de los complejos inmunes de IgA mesangial es un subtipo morfológico de la nefritis lúpica. Sin embargo, la ausencia de otras inmunoglobulinas, en particular IgG, y de las fracciones del complemento de la vía clásica, como C1q y C4, están fuertemente en contra de este diagnóstico.
Sin la historia clínica, el diagnóstico histológico de la nefritis lúpica no se habría considerado, e incluso con el LES clínico establecido, los hallazgos histológicos no justifican el diagnóstico de nefritis lúpica ni siquiera de forma atípica.
En nuestro paciente, la ausencia de cualquier etiología reconocida justifica la clasificación de IgAN primaria o idiopática. Dada la alta frecuencia de IgAN, esta asociación podría ser meramente casual.
Otro punto interesante es que nuestra paciente presentó anticuerpos anti-Ro / SSA positivos y ANA persistentemente negativos. La asociación entre anti-Ro y la enfermedad renal en el LES sigue siendo la enfermedad renal en los pacientes con LES puede asociarse inversamente con la presencia de este factor serológico 13. De hecho, los anticuerpos anti-Ro / SSA se encuentran principalmente en pacientes con LES y síndrome de Sjogren 13,14. Los anticuerpos anti-Ro / SSA se encuentran en aproximadamente 10 a 60 por ciento de los pacientes con LES, y la prevalencia depende de la metodología empleada 14. Estos anticuerpos se han asociado con fotosensibilidad, una erupción conocida como lupus cutáneo subagudo, vasculitis cutánea, enfermedad pulmonar intersticial, enfermedad neonatal y bloqueo cardíaco congénito 14. Sin embargo, en raras ocasiones, la prueba de anticuerpos anti-Ro / SSA puede ser útil para sugerir un diagnóstico de enfermedad autoinmune sistémica frente a unos ANA negativos 14.
A pesar de sus fascinantes implicaciones biológicas y su evidente importancia clínica, las nefritis sin lupus observadas en pacientes con LES siguen siendo poco conocidas y limitadas a informes de casos aislados15. Los casos reportados en la literatura 1,8,9 y lo nuestro sugieren que las enfermedades renales distintas de la nefritis por lupus deben sospecharse clínicamente en pacientes con remisión serológica y clínica de la actividad del lupus que presenten anomalías renales.
Nuestro caso destaca la importancia de la biopsia renal en pacientes con lupus, ya que un diagnóstico correcto permitirá utilizar el tratamiento más adecuado, evitando regímenes inmunosupresores innecesarios.
DISCUSSION
IgAN has previously been described in association with various autoimmune diseases, but to the best of our knowledge, few previous cases of IgAN associated with SLE have been reported 1,8,9. Despite being distinct diseases, with different clinical and pathological findings, and with different prognosis, they share some common physiopathological characteristics 1,8,9.
Renal involvement in SLE is characterized by variable grades of proteinuria, urinary sediment changes with micro-hematuria and erythrocyte casts, hypertension and progressive renal dysfunction.
Typical extra-renal signs in the skin, joints and blood are often associated with LN, which usually permit easy differentiation from IgAN. Nevertheless, in IgAN there may also be some extra-renal features, such as arthralgia, vasculitic skin lesions and erythema result in confusion with SLE 9,10.
IgAN is the most common cause of idiopathic glomerulonephritis in the world 11. Although this disorder was initially thought to have a benign course, it is now recognized that slow progression to end stage renal disease occurs in up to 50 percent of affected patients 11. The pathogenesis of IgAN is uncertain and its etiology is unknown. Abnormal immune regulation results in formation of IgA containing immune complexes characterized by their high affinity for the mesangium 5.
SLE is clearly an autoimmune disease; attempts to relate IgAN to autoimmunity have failed due to a lack of significant auto-antibodies in IgAN 12.
The immuno-histological aspects of IgAN are characterized by mesangial proliferation and mesangial deposits of IgA1 (usually absent in lupus nephritis), C3 fraction of complement and occasionally IgG and IgM, which are responsible for complement activation with the consequential release of inflammatory mediators 5. Although the clinical picture can be highly suggestive, the diagnosis can only be confirmed by a kidney biopsy.
Our patient had characteristic clinical features of SLE, with five diagnostic criteria of the American College of Rheumatology. To the best of our knowledge, our patient had no family history suggestive of an inherited condition such as immunodeficiency.
Two years after the clinical diagnosis of SLE, renal involvement (mild proteinuria and microscopic hematuria with a well-preserved renal function) was detected, and the patient underwent renal biopsy. In our case the kidney biopsy showed no typical histological findings of SLE.
One may argue that the predominance of mesangial IgA immune complexes is a morphological subtype of lupus nephritis. However, the absence of other immunoglobulins, in particular IgG, and of complement fractions of the classical pathway, such as C1q and C4, are strongly against this diagnosis.
Without the clinical history, the histological diagnosis of lupus nephritis would not have been considered, and even with established clinical SLE, the histological findings do not justify a diagnosis of lupus nephritis even of an atypical form.
In our patient the absence of any recognized etiology justifies the classification of primary or idiopathic IgAN. Given the high frequency of IgAN these association could be merely casual.
Another interesting point is that our patient presented with positive anti -Ro / SSA antibodies and persistently tested negative for ANA. The association between anti-Ro and renal disease in SLE remains renal disease in SLE patients may be inversely associated with the presence of this serological factor13. In fact, anti-Ro/SSA antibodies are primarily found in patients with SLE and Sjogren’s syndrome 13,14. Anti-Ro/SSA antibodies are found in approximately 10– 60 percent of patients with SLE, and the prevalence depends upon the methodology employed 14. These antibodies have been associated with photosensitivity, a rash known as subacute cutaneous lupus, cutaneous vasculitis, interstitial lung disease, neonatal disease and congenital heart block 14. Nevertheless, on rare occasions, the anti-Ro/SSA antibody test may be useful in suggesting a diagnosis of systemic autoimmune disease in face of a negative ANA14.
In spite of their fascinating biological implications and obvious clinical significance, non-lupus nephritides seen in patients with SLE remain poorly understood and limited to isolated case reports 15. The cases reported in the literature 1,8,9 and our own suggest that renal diseases other than lupus nephritis should be clinically suspected in patients with serological and clinical remission of lupus activity who are found to have renal abnormalities.
Our case highlights the importance of renal biopsy in lupus patients, since a correct diagnosis will permit the most appropriate treatment to be used, avoiding unnecessary immunosuppressive regimens.
Declaracion de conflicto de interés:
Ninguno declarado.
Conflict of interest statement:
None declared.
ESTUDIO MULTICÉNTRICO DE PREVALENCIA DE ENFERMEDAD RENAL CRÓNICA
Autores:
Aymard, AL
Vanden Ryn
RM; Aranda, C
Suárez, R; Piaggio
N; Simesen, G
Bearzi, L
Lejtman, N
Chávez, C
Pinheiro, M
Correspondencia:
Dr. Adrián L. Aymard
Salguero 560 2º Piso Laboratorio
1171 CABA, Argentina.
011 4860 1000. Int: 5125
E-mail: adrianaymard@hotmail.com
Resumen:
Introducción: La enfermedad renal crónica es secundaria a múltiples etiologías y se caracteriza por ser silente, siendo reconocido su bajo índice de diagnóstico precoz. El objetivo del trabajo fue evaluar la prevalencia de enfermedad renal crónica, según las guías KDIGO, en pacientes que presentaron algún factor de riesgo en las diferentes regiones del país con participación de los laboratorios asociados a la red ALAC.
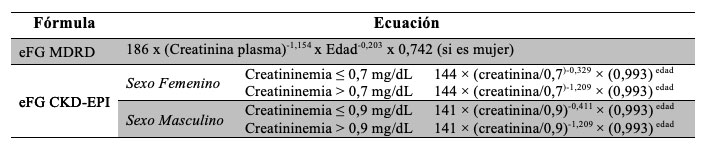
Métodos: Se incluyeron pacientes entre 18-70 años que presentaron algún factor de riesgo asociado. Se obtuvieron 2685 muestras de 30 laboratorios participantes durante Septiembre 2015-Abril 2016. Se midió Relación Albúmina-Creatinina (RAC) y se estimó el filtrado glomerular (eFG) a través de las fórmulas MDRD-4 y CKD-EPI (Roche Modular P).
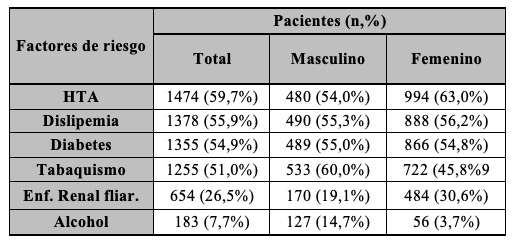
Resultados: El 64% fueron mujeres y el 36% varones. El índice de masa corporal (IMC) arrojó un 73,4% de los pacientes con sobrepeso u obesidad. La prevalencia de factores de riesgo fue: hipertensión 59,7%; dislipemias 55,9%, diabetes 54,9%, tabaquismo 51,0%, antecedentes renales 26,5%, consumo de alcohol 7,7%. La prevalencia de RAC alterado fue 7,5%; y de eFG menor a 60 mL/min/1,73m2 fue 8,1% según MDRD-4 y 4,9% según CKD-EPI. El cuadro de severidad de riesgos muestra que un 14,4% de pacientes presentan algún riesgo de ERC utilizando MDRD-4 y 11,2% si se utiliza CKD-EPI.
Conclusiones: Se demostró la alta prevalencia de alteraciones de la función renal en pacientes ambulatorios que concurrieron a consultas de atención primaria. Su detección temprana permitió su atención especializada con la intención de mejorar la calidad de vida de nuestros pacientes.
Palabras clave: enfermedad renal crónica; relación albúmina/creatinina; factores de riesgo asociados; estimación del filtrado glomerular.
ABSTRACT:
Background: Chronic kidney disease (CKD) is secondary to multiple etiologies and it is characterized by being silent and having a low rate of early diagnosis. The objective of this study was to evaluate the prevalence of CKD, according to the KDIGO guidelines, in patients who showed risk factors in the different regions of the country with the participation of the laboratories associated with the ALAC network.
Methods: Patients between 18-70 years old who showed associated risk factors were included 2685 samples were obtained from 30 participating laboratories during September 2015-April 2016. The albumin/creatinine ratio (ACR) was measured (Roche Modular P) and the glomerular filtration rate was estimated (eGFR) through the MDRD-4 and CKD-EPI formulas.
Results: 64% of participants were women while 36% were men. The body mass index (BMI) showed that 73,4% of the patients were overweight or obese. The prevalence of risk factors was: hypertension 59,7%; dyslipidemia 55,9%, diabetes 54,9%, tobacco smoking 51,0%, previous medical history of renal disease 26,5%, alcoholism 7,7%. The prevalence of altered ACR was 7,5%; and of eGFR lower than 60mL/min/1,73m2 was 8,1% according to MDRD-4 and 4,9% according to CKD-EPI. The severity of risk chart illustrates that 14,4% of the patients show some risk of CKD using MDRD-4 and 11,2% if CKD-EPI is used.
Conclusions: A high prevalence of alterations in the renal function was showed in outpatients who attended primary healthcare consultations. The early detection of the disease enabled its specialized attention with the intention of improving the quality of life of patients.
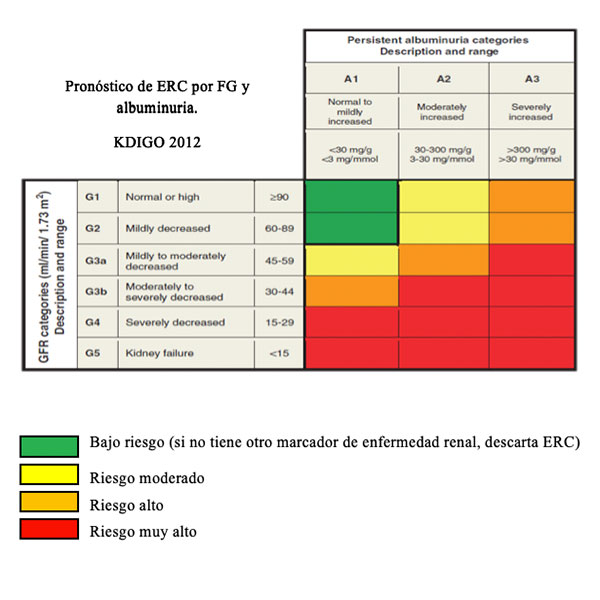
La enfermedad renal crónica (ERC) es definida por la presencia de un daño renal durante un período de al menos tres meses, que se evidencia por alteraciones en su función o por la presencia de marcadores específicos. 1,2,3 Es una entidad clínica secundaria a diversas etiologías caracterizada por ser silente o presentar síntomas inespecíficos en sus estados iniciales. Este es el motivo por el cual, a pesar de contar con estrategias de efectividad probada para su detección, frecuentemente no es reconocida de manera precoz, sino hasta los estadios más avanzados que requieren tratamientos sustitutivos o incluso trasplante renal, con la carga de morbilidad acompañante, el deterioro de la calidad de vida del paciente y costos elevados. 4 Su cronicidad no es sinónimo de irreversibilidad, ya que en algunos casos ERC es reversible de forma espontánea o a través del tratamiento y en otros casos la terapia puede causar una regresión parcial del daño renal, mejorando la funcionalidad. 2,3 Sus dos causas más prevalentes en el mundo son la Diabetes Mellitus y la Hipertensión arterial, y su complicación más severa y frecuente es la enfermedad cardiovascular (ECV). Este es el motivo por el cual en la actualidad se concentran los esfuerzos en su detección temprana, donde la mayoría de las veces presenta un bajo índice de diagnóstico. 5 Por todo esto, en el desarrollo de programas de detección precoz y tratamiento de enfermedades renales es esencial el uso de herramientas simples y seguras para la evaluación de la función renal. La ERC puede clasificarse según las guías KDIGO 2012, considerando la estimación del filtrado glomerular (eFG) y los valores de albuminuria del paciente, quedando definidos cuatro grupos: pacientes con bajo riesgo (si no presentan otro marcador de enfermedad renal, descartan ERC); pacientes con riesgo moderado; pacientes con riesgo alto; y pacientes con riesgo muy alto. 2,3 Una de las estrategias de prevención de las complicaciones asociadas a la insuficiencia renal es conocer el grado de la función renal, valoración que habitualmente se realiza con la creatinina sérica. La creatinina es un buen marcador de seguimiento pero no lo es del filtrado glomerular, ya que valores normales o ligeramente elevados no se corresponden en muchas ocasiones con la severidad del cuadro. Ante esta dificultad, las sociedades profesionales y científicas nacionales recomiendan la evaluación de la función renal a través de la estimación del índice de filtrado glomerular (eFG) por diferentes ecuaciones, que incluyen los factores sexo, edad y raza. La forma más utilizada en los pacientes adultos es la Modification of Diet in Renal Disease (MDRD-4). Recientes opiniones evalúan la importancia de utilizar la fórmula Chronic Kidney Disease Epidemiology Collaboration (CKD-EPI), en particular cuando la afección de la función renal es leve. El principal motivo de esta discusión se basa en las limitaciones que presenta la fórmula MDRD-4 derivadas a partir de que su estudio se desarrolló en personas con enfermedad renal crónica y de este modo sus principales objeciones radican en la imprecisión y la subestimación sistemática, sobre todo en valores de eFG mayores a 90 mL/min/1,73 m2. La fórmula del grupo CKD-EPI fue desarrollada a partir de una población de 8254 individuos que incluye como variables a la creatinina sérica, la edad, la raza y el sexo, obteniéndose diferentes versiones según la etnia, el sexo y el valor de creatinina. Sus resultados, según indican los autores, son más exactos y precisos, en especial para valores de eFG mayores de 60 mL/min/1,73 m2. 6,7,8,9,10
El objetivo del trabajo fue estudiar la prevalencia de enfermedad renal crónica, utilizando las recomendaciones de las guías KDIGO 2012, en pacientes que presentaron algún factor de riesgo. Se evaluaron y compararon los resultados de las fórmulas recomendadas, analizando su concordancia.
MÉTODOS:
Se seleccionaron pacientes entre 18-70 años de edad que presentaron algún factor de riesgo asociado y concurrieron de forma voluntaria a los laboratorios de ALAC participantes. Estos fueron los que difundieron, sensibilizaron e invitaron al público en general a través de afiches que destacaron la importancia de la pesquisa de esta enfermedad.
Criterios de inclusión: Se incluyeron aquellos pacientes que presentaron al menos uno de los siguientes factores de riesgo: Diabetes Mellitus (personal o familiar), Hipertensión arterial (siempre que no se encuentren medicados con inhibidores de la enzima convertidora de angiotensina-IECA o antagonistas del receptor de angiotensina II-ARA II antihipertensivos que a la vez son renoprotectores), antecedentes de infarto agudo de miocardio (IAM) o de accidente cardiovascular (ACV), tabaquismo actual o pasado, consumidores de drogas nefrotóxicas, antecedentes de patologías obstructivas urológicas, o de enfermedad renal familiar, antecedentes de enfermedades sistémicas que afectan al riñón como Lupus Eritematoso Sistémico, consumo de alcohol, antecedentes de dislipemia, y/o aquellos en los que se detectó proteinuria o hematuria. 11,12
Criterios de exclusión: pacientes que no presentaron factores de riesgo, embarazadas, actividad física realizada en las últimas 72 hs, estados febriles o infecciones del tracto urinario en los siete días previos, pacientes que estuvieran en los dos días previos o tres posteriores al ciclo menstrual. 12,13
Cada paciente incluido firmó el respectivo consentimiento informado.
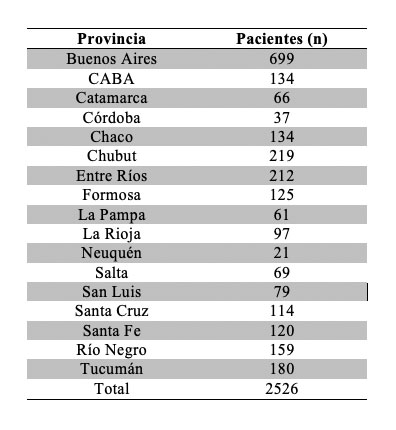
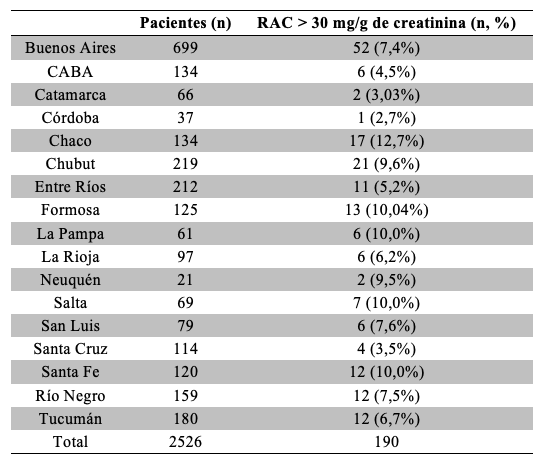
Cada laboratorio designó agentes sanitarios referentes para orientar a los interesados en participar utilizando una encuesta modelo que permitió incluir o excluir a los participantes a la vez que brindó información para su posterior clasificación. A partir de los datos de peso y altura se utilizó la fórmula Peso(kg)/Altura2(m2) para obtener el índice de masa corporal (IMC), utilizando la definición de la Organización Mundial de la Salud (OMS). 14 Participaron 2526 pacientes de 30 Laboratorios de todo el país Tabla 1, durante los meses de Septiembre de 2015 hasta Abril de 2016. El 64% de los participantes fueron mujeres, con una media de edad de 47 + 14 años, mientras el 36% restante fueron varones con una media en años de 48 + 13.
Se recolectó primera orina de la mañana (10 mL) para medir Relación Albúmina-Creatinina (RAC) y suero (2 mL) para medir Creatinina y estimar el filtrado glomerular (eFG) a través de las fórmulas MDRD-4 y CKD-EPI Tabla 2. La Albúmina se midió por inmunoturbidimetría ALB 2, Roche, y la Creatinina por el método Jaffé compensado Creatinina, (valores de referencia: hombres 0,7-1,2 mg/dL; mujeres: 0,5-0,9 mg/dL), en el equipo automatizado Roche Modular P, Roche. Se utilizaron Controles de Calidad Internos Biorad y Roche; y Controles de Calidad Externos: CEMIC-Progba, PEEC y DGKL. Las muestras se procesaron en un único Centro efector (IACA Laboratorios), que comunicó los resultados de manera sistemática. Con los datos obtenidos se confeccionó el cuadro de clasificación de riesgos recomendado por las guías KDIGO 2012 Cuadro 2. Los pacientes con muestras con resultados alterados para RAC (mayores a 30 mg/g de Creatinina) fueron contactados para repetir la determinación luego de tres meses a partir de la primera cita para su confirmación. En caso de dos resultados alterados, se recomendó al paciente asistir a una consulta con un médico especialista. En caso de no confirmarse el primer resultado se le sugirió repetir este estudio en seis meses de acuerdo al criterio de su médico.
No deben ser usadas en menores de 18 años ni en mayores de 70, embarazadas, pacientes con enfermedades consuntivas (TBC, HIV, cáncer, desnutrición, etc.) que producen delgadez extrema o personas con tamaños corporales, masa muscular o estatus nutricionales extremos 5.
En el presente trabajo no se ha utilizado un método patrón de medición del filtrado glomerular, de modo que los resultados considerados positivos o negativos que definen una mayor o menor clasificación de pacientes por estadio, se basan en la bibliografía estudiada.
Análisis estadístico: se aplicó el Test de Student para muestras Independientes y Análisis de la Varianza (ANOVA). Las variables categóricas se compararon mediante la prueba de Chi-cuadrado con corrección por continuidad de Yates. Cuando se compararon más de dos grupos se aplicó la corrección de Bonferroni al nivel de significación. Para estudiar los dos métodos de evaluación de insuficiencia renal (eFG MDRD-4 y eFG CKD-EPI) se aplicó Test de Student para muestras relacionadas, Correlación lineal de Pearson, Regresión lineal de Pearson. El análisis de concordancia se realizó con el coeficiente de correlación intraclase y el método de regresión de Passing y Bablok. En todos los casos los test estadísticos aplicados fueron para muestras independientes y con un nivel de significación menor del 5%.Todos los análisis estadísticos se realizaron utilizando los programas informáticos SPSS (versión 20), STATA 12, MS Excel 2013, MedCalc; Epidat 4.0.
RESULTADOS:
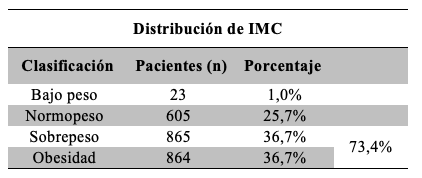
A partir de los datos recolectados en la Encuesta se calculó el IMC. El 73,4% de los pacientes presentó sobrepeso u obesidad al momento de su participación Tabla 3.
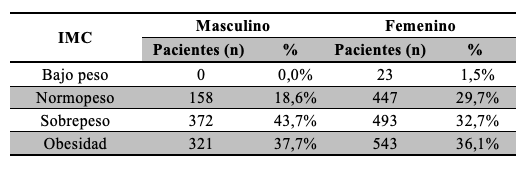
Teniendo en cuenta el sexo de los participantes la distribución de los datos de IMC se describió en la Tabla 4.
Del análisis de la encuesta surgió la prevalencia de los factores de riesgo en la población participante: hipertensión fue el más frecuente con 59,7%; dislipemia 55,9%, diabetes 54,9%, tabaquismo 51,0%, antecedentes de patologías renales 26,5%, y consumo de alcohol 7,7% Tabla 5.
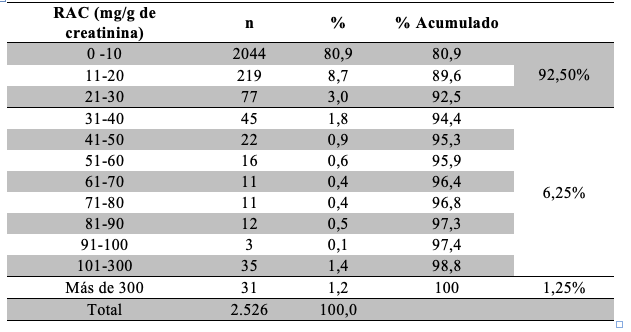
De los resultados obtenidos a partir del cálculo de RAC surgió que 7,5% de los pacientes presentaron valores alterados. Dentro de este grupo, se establecieron dos puntos de corte para evaluar la severidad de los resultados de RAC y se observó que 6,25% tuvieron valores entre 30 y 300 mg/g de creatinina y 1,25% mayores de 300 mg/g de creatinina. En la Tabla 6 se observa la frecuencia de resultados de RAC evaluados en rangos de a 10 unidades (mg/g de creatinina).
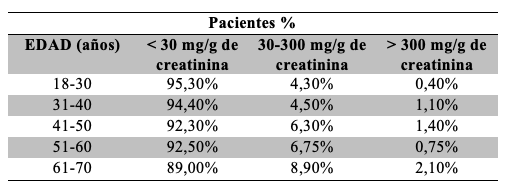
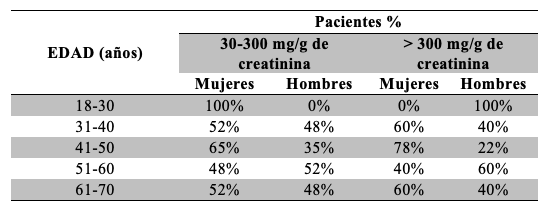
Se observó que el porcentaje de pacientes que participaron del estudio aumenta de acuerdo a la edad, de modo que el 71% de los pacientes incluido supera los 40 años Tabla 7. Se observó un incremento del porcentaje de resultados de RAC alterados conforme aumenta la edad de los pacientes, considerando rangos de 10 años Tabla 8. Respecto de la relación sexo-edad, el porcentaje de mujeres con valores de RAC alterados se mantuvo superior al de los hombres en casi todos los rangos Tabla 9.
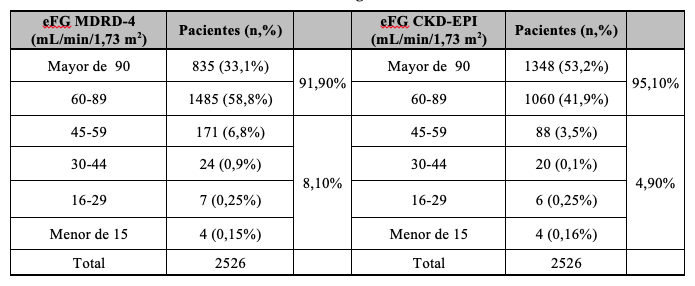
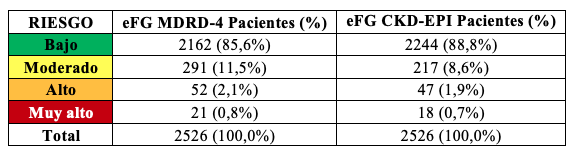
Los resultados de la eFG mostraron que aplicando la fórmula MDRD-4 un 8,1% de los pacientes presentaba valores menores a 60 mL/min/1,73m2 (considerado el límite inferior de referencia). Utilizando la ecuación CKD-EPI el porcentaje de pacientes por debajo de este límite fue 4,9%, de modo que 3,2% de los pacientes se reclasificarían a valores de eFG superiores a 60 ml/min/1,73m2 y no deberían considerarse con función renal alterada Tabla 10.
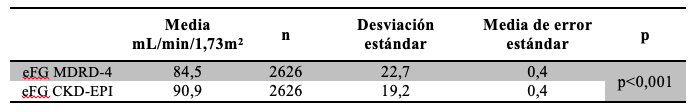
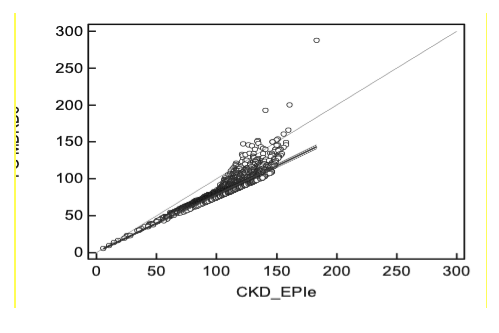
De la comparación de fórmulas surgió que existe una correlación (Pearson) significativa entre ambas: 0,899. De la evaluación de la concordancia resultó que eFG MDRD-4 presenta valores más bajos respecto de eFG CKD-EPI, en especial en el rango de los resultados mayores a 90 mL/min/1,73m2. La diferencia entre ambas fórmulas MDRD-4 vs CKD-EPI tuvo una media igual a - 6,44 Tabla 11 y Tabla 12; Gráfico 1, siendo esta diferencia estadísticamente significativa. (p< 0,01).
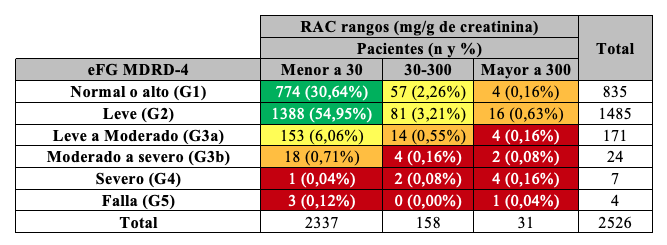
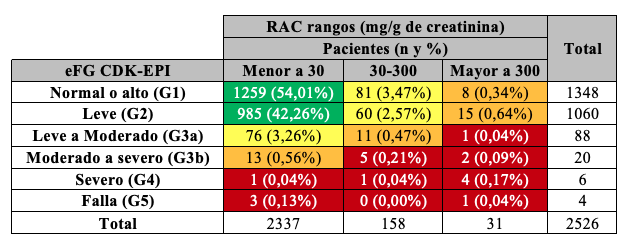
Según las guías KDIGO 2012 se clasificó la población en estadios de ERC (G1 a G5) tomando como referencia el valor de eFG, y subdividiendo el estadio 3 en 3a y 3b, siendo éste el valor crítico de decisión ya que implica eFG entre 60 y 30 mL/min/1,73 m2. A partir de los resultados de RAC y eFG, se definió el cuadro de severidad de riesgos resultante. Aplicando MDRD-4 se observó que 14,4% de los participantes presentaban al menos un nivel riesgo de ERC. Si se utiliza CKD-EPI, 11,2% de los pacientes presentaron algún tipo de riesgo. De esta manera, se observó que utilizando la ecuación CKD-EPI el número de pacientes con riesgo de desarrollar ERC disminuyó, reclasificando pacientes (3,2%) hacia categorías de riesgo más favorables Tablas 13, 14, 15.
Del total de pacientes evaluados, 190 presentaron un valor de RAC mayor a 30 mg/g de creatinina. Este grupo fue recitado luego de un período de al menos tres meses, respondiendo al contacto para su reevaluación 91 pacientes. De estos pacientes, 65 confirmaron el resultado elevado de RAC y 26 tuvieron un resultado inferior a 30 mg/g de creatinina en la segunda muestra, por lo que se consideraron negativo para el estudio. Los 99 pacientes que no concurrieron presentaron diversas causas que justificaron su ausencia: algunos iniciaron tratamiento con su propio médico, otros abandonaron el protocolo no respondiendo al llamado y un paciente falleció en ese período.
Las distintas provincias del país, representadas por los Laboratorios participantes, presentaron un porcentaje de pacientes con RAC alterado que tiene su máximo en Chaco con 12,7% Tabla 16.
En los 65 participantes que confirmaron el valor alterado de RAC, se evaluó la distribución de los factores de riesgo asociados: los más prevalentes fueron Diabetes (58,2%) y Sobrepeso/Obesidad (56,0%), seguido de Hipertensión Arterial, Dislipemia y Tabaquismo (40,6%, 39,6% y 39,6% respectivamente).
DISCUSIÓN:
En este estudio multicéntrico, las personas mayores de 40 años fueron las de mayor participación. Es importante tener en cuenta que la edad es un factor de riesgo independiente para enfermedad renal crónica, ya que el número de glomérulos escleróticos aumenta a partir de los 50 años debido a isquemia glomerular secundaria a cambios en el flujo sanguíneo renal producidos por el envejecimiento. También el número de mujeres participantes fue superior al de los hombres, lo que podría explicarse por la mayor asistencia de las mujeres a las instituciones de salud y su mayor apego a la prevención sanitaria 15, 16, 17. En cuanto a los distintos factores de riesgo asociados a ERC, el antecedente de hipertensión arterial fue el más frecuente en nuestros pacientes, siendo importante resaltar que diabetes mellitus e hipertensión arterial son a nivel mundial las causas principales de ERC 8, 18. Cabe señalar también que la obesidad es un factor de riesgo asociado a microalbuminuria ya que los riñones de estos individuos muestran cambios histológicos similares a los observados en enfermedad renal diabética.
Diferentes estudios observacionales a nivel mundial, estiman la prevalencia global de ERC entre 11-13%, con resultados que varían ampliamente entre los estudios presentando alta heterogeneidad. Este trabajo presenta resultados algo menores, pero que demuestran la existencia de un porcentaje elevado de pacientes afectados que desconocen su condición. 19, 20
La utilización de nuevas ecuaciones para valorar la eFG implica su validación en poblaciones de distintas características clínicas. Este estudio comparó un importante número de pacientes, abarcando un amplio rango de valores de eFG, con una distribución geográfica extendida en todo el país, y con características de atención ambulatoria e intención preventiva. 21
Los resultados de este estudio aportan información sobre ERC en una población distribuida de manera relativamente uniforme a lo largo del país. Con los resultados obtenidos se aconsejó y derivó a los pacientes comprometidos para la consulta con especialistas y el seguimiento de su probable enfermedad. Una de las limitaciones del estudio fue que no permitió discriminar una disminución transitoria de la función renal de una ERC establecida en todos los casos, considerando que no todos los pacientes repitieron los exámenes luego de tres meses. Esto puede mitigarse teniendo en cuenta que la información aportada a los pacientes y sus médicos permitió que en la mayoría de los casos se tomaran medidas acordes aún cuando salieron del programa.
CONCLUSIÓN:
En este trabajo se ha intentado avanzar en el conocimiento de la prevalencia de enfermedad renal crónica en pacientes que voluntariamente aceptaron participar en los laboratorios asociados a la red ALAC. La distribución geográfica de la asociación, permitió realizar el estudio a lo largo de todo el país. Este trabajo demuestra que un porcentaje elevado de pacientes ambulatorios presentan signos de sospecha de alteraciones en su función renal, con valores en la relación albúmina/creatinina urinarias y estimación de filtrado glomerular alterados. Sería adecuado asociar a cada pedido médico de creatinina sérica el cálculo de la ecuación de eFG, para completar de manera más segura la evaluación inicial de la funcionalidad renal. Estos resultados deberán siempre ser ratificados con otros parámetros como RAC y repeticiones en el transcurso de tres meses.
La importancia de este estudio radica en que siendo estas alteraciones metabólicas silentes su hallazgo temprano a nivel de la atención primaria permite evitar futuras complicaciones y su posterior referencia a especialistas que logren mejorar la calidad de vida a largo plazo. Estas complicaciones pueden limitar el bienestar de estos pacientes, fundamentalmente en su función cardiovascular. La participación de Laboratorios nucleados bajo la red de Asociación de Laboratorios de Alta Complejidad de todo el país, concentrados en un objetivo común, brinda herramientas accesibles y de bajo costo para la prevención y asistencia temprana de la salud, en busca de la seguridad, el cuidado y la educación del paciente modificando factores de riesgo asociados.
CONFLICTO DE INTERÉS:
Los autores declaran no tener ningún conflicto de intereses.
AGRADECIMIENTOS:
Los autores agradecen la participación voluntaria de los pacientes y los aportes del Dr. Luis Sintado (Jefe del Servicio de Nefrología del Hospital Durand- CABA) por su asesoramiento médico, del licenciado Pablo Salgado por su contribución en el análisis estadístico, a los Laboratorios colegas de ALAC: LACE- Laboratorios de Análisis Clínicos Especializados (Provincia de Córdoba),Laboratorio de Análisis Clínicos Hematológicos y Banco de Sangre LACHyBS (Provincia de Neuquén), IDAC SA (Provincia de Río Negro), Laboratorios de Alta Complejidad Dres Castagnino (Provincia de Bs As), Instituto de Bioquímica Clínica S.R.L. IBC (Provincia de Santa Fe), por su participación y colaboración, y a las empresas Roche y Wiener Lab que ofrecieron sus reactivos para la realización de este trabajo.
Este estudio ha recibido el apoyo de la Asociación de Laboratorios de Alta Complejidad (ALAC) y de la Fundación ALAC, así como el compromiso y dedicación de todos los Laboratorios participantes a través del grupo ERC-ALAC.
NEFRITIS LÚPICA MEMBRANOSA: REPORTE DE UN CASO
MEMBRANOUS LUPUS NEPHRITIS: A CASE REPORT
Autores:
Moreira, T.
Rodrigues dos Santos, L.
Vasconcelos, G.
Silva Cruz, M.
Da Cruz, M.
Palma, N.
Batista, V.
Meireles, R.
Botelho, C.
Correspondencia:
Dra. Teresa Moreira Internal Medicine Department, Centro Hospitalar do Tâmega e Sousa, Penafiel, Portugal. teresa_margaridam@hotmail.com Avenida do Hospital Padre Américo 210, 4564-007 Guilhufe 00351915575343
Resumen:
La enfermedad renal es la mayor causa de morbimortalidad en el Lupus Eritematoso Sistémico (LES). Entre las clases histológicas de la nefritis lúpica, la Nefritis Lúpica Membranosa (NLM) representa sólo un quinto de todos los casos, siendo la inmunofluorescencia fundamental para el diagnóstico.
El pronóstico de la NLM, a largo plazo es incierto, con un porcentaje significativo progresando hacia la enfermedad renal crónica estadio 5D (10 a 20%).
La evidencia existente sobre la eficacia relativa de las diferentes terapias es limitada.
Los autores presentan el caso clínico de un hombre en la 6ª década de vida con biopsia renal a revelar NLM, en ausencia de otras manifestaciones clínicas y de laboratorio del LES. También discuten el tratamiento efectuado, presentando el posterior seguimiento.
Palabras clave: nefritis lúpica membranosa, síndrome nefrótico, micofenolato de mofetilo
ABSTRACT
Renal disease is the major cause of morbidity and mortality in systemic lupus erythematosus (SLE). Among the histological classes of lupus nephritis, Membranous Lupus Nephritis (MLN) represents only one fifth of all cases, with immunofluorescence being fundamental for diagnosis.
The long-term prognosis of MLN is uncertain, with a significant percentage progressing to chronic renal disease stage 5D (10 to 20%).
There is limited evidence of the relative effectiveness of different therapies.
The authors present the clinical case of a man in the 6th decade of life with renal biopsy revealing NLM, in the absence of other clinical and laboratory manifestations of SLE. They also discuss the treatment carried out, presenting the subsequent follow-up.
El LES es una enfermedad multisistémica mediada inmunológicamente, con una amplia variedad de manifestaciones clínicas. El inicio del LES en pacientes >60 años es bastante inusual. 1,2
La Nefritis Lúpica Membranosa (NLM) o clase V, afecta entre 10 y 20% de los pacientes con manifestaciones renales del Lupus Eritematoso Sistémico (LES). A diferencia de las clases III y IV, surge frecuentemente en ausencia de otras manifestaciones clínicas y de laboratorio del LES, dificultando el diagnóstico diferencial con la Nefropatía Membranosa Idiopática (NMI). Como la NMI, se presenta habitualmente con síndrome nefrótico y con un patrón histológico indistinguible de la forma idiopática.3 La inmunofluorescencia es fundamental para el diagnóstico.4
El pronóstico de la NLM a largo plazo es incierto, con un porcentaje significativo a progresar a la enfermedad renal crónica estadio 5D (10 a 20%). El riesgo cardiovascular y tromboembólico es alto.5 La historia natural de la enfermedad y los factores de riesgo para la progresión no están bien establecidos, lo que dificulta la decisión sobre el inicio y el tipo de terapia a instituir.6
Las medidas anti-proteinúricas, agentes alquilantes, inhibidores de la calcineurina, micofenolato de mofetilo y rituximab son algunas de las opciones terapéuticas. La evidencia existente sobre la eficacia relativa de las diferentes terapias es limitada.7
INTRODUCTION
SLE is an immunologically mediated multisystemic disease with a wide variety of clinical manifestations. The onset of SLE in patients> 60 years is quite unusual. 1,2
Membranous Lupus Nephritis (MLN) or Class V affects between 10% and 20% of patients with renal manifestations of Systemic Lupus Erythematosus (SLE). Unlike classes III and IV, it frequently arises in the absence of other clinical and laboratory manifestations of SLE, making difficult differential diagnosis with Idiopathic Membranous Nephropathy (IMN). Like IMN, it usually presents with nephrotic syndrome and with a histological pattern indistinguishable from the idiopathic form. 3 Immunofluorescence is essential for diagnosis. 4
Long-term prognosis of MLN is uncertain, with a significant percentage progressing to chronic renal disease stage 5D (10 to 20%). Cardiovascular and thromboembolic risk are high. 5 The natural history of the disease and the risk factors for progression are not well established, making difficult to decide on the onset and type of therapy to be instituted. 6
Antiproteinuric measures, alkylating agents, calcineurin inhibitors, mycophenolate mofetil and rituximab are some of the therapeutic options. There is limited evidence of the relative effectiveness of different therapies.7
REPORTE DE CASO
Hombre, caucásico, 65 años. Diabético, hipertenso con dislipidemia. Edema bilateral de los miembros inferiores de inicio súbito de agravamiento progresivo con cerca de 4 meses de evolución. Astenia, anorexia y disminución del débito urinario progresivos desde hace 3 días.
En el examen clínico con edema bilateral marcado de los miembros inferiores. Los análisis de sangre de laboratorio revelaron un aumento de la tasa de sedimentación de eritrocitos (TSH 88mm/h), anemia normocrómica normocítica (Hbg: 10,5 g/dl), ligera retención azotada (urea: 97mg/dl; Cr: 2,2mg/dL), hipoalbuminemia grave (1,3 g/dl) y dislipidemia (CT: 433mg/dL) c-LDL: 311 mg/dl). La excreción de proteínas en orina de 24 horas fue de 24g.
El análisis de orina fue positivo para proteínas, glucosa y sangre.
Disminución de IgG (< 200mg/dL). Las fracciones de C3 y C4 del complemento sérico son normales. Los títulos de ANA y anti-dsDNA son indetectables. Cadenas ligeras séricas y urinarias normales.
ASLO negativo. Ausencia de crioglobulinas circulantes.
Serologías VHB, VHC y VIH negativas. Marcadores tumorales negativos. Ecografía renal con riñones normales. La tomografía computarizada toracoabdominal-pélvica, la endoscopia digestiva superior e inferior fueron normales.
Se realizó una biopsia renal percutánea con aguja. El examen histológico a la luz mostro 6 glomérulos con engrosamiento de la membrana basal; tinción de plata con espículas en la membrana basal glomerular Figura 1; tinción de inmunofluorescencia con depósitos granulares y gruesos de C3, C4, C1q, IgA, IgG e IgM Figura 2. Se estableció el diagnóstico de glomerulonefritis membranosa con depósitos de inmunocomplejos - glomerulonefritis lúpica clase V.
Se inició pulsos de metilprednisolona (1g durante 3 días), prednisolona 0,5mg/kg/día (40mg) y micofenolato de mofetilo en esquema de aumento progresivo.
Después de 48 meses del inicio de la terapia, con prednisolona 2.5mg id y hidroxicloroquina 400mg id, clínicamente bien, sin edema de los miembros inferiores; normalización de la función renal; disminución progresiva de proteinuria, actualmente con 0,42gr/24h; sedimento urinario inactivo. Complemento, ANA y anti-dsDNA sin alteraciones. Sin complicaciones relacionadas con la terapia inmunosupresora.
CASE REPORT
A 65-year-old, caucasian man, diabetic, hypertensive with dyslipidemia was referred to our clinic with progressive sudden onset bilateral edema of the lower limbs with about 4 months. Progressive asthenia, anorexia and decreased urine output since 3 days ago. Clinical examination revealed marked bilateral edema of the lower limbs.
Laboratory blood tests revealed an increase in erythrocyte sedimentation rate (ESR 88 mm/1sth), normocytic normochromic anemia (Hb: 10.5g/dL), slight nitrogen retention (urea: 97mg/dL, SCr:2.2mg/dL), severe hypoalbuminemia (1.3 g/dL) and dyslipidemia (TC: 433 mg/dL, C-LDL: 311mg/dL). 24-hour urine protein excretion was 24g.
The urine analysis was positive for protein, glucose and blood.
Decreased IgG (< 200mg/dL). Serum complement C3 and C4 fractions are normal. ANA and anti-dsDNA titers are undetectable. Normal serum and urinary light chains.
Negative ASLO. Absence of circulating cryoglobulins.
HBV, HCV and HIV negative serology. Negative tumor markers. Renal ultrasound with normal kidneys. Thoracoabdominal-pelvic CT scan, upper and lower gastrointestinal endoscopy were normal.
A percutaneous needle kidney biopsy was performed. Histological examination at light showed 6 glomeruli with basement membrane thickening; silver staining with spicules in the glomerular basement membrane Figure 1; immunofluorescence staining with granular and coarse deposits of C3, C4, C1q, IgA, IgG and IgM Figure 2. The diagnosis of Membranous glomerulonephritis with immunocomplexes deposits - Class V Lupus Glomerulonephritis was established.
Started pulses of methylprednisolone (1g for 3 days), prednisolone 0.5mg/kg/day (40mg) and mycophenolate mofetil in a progressive increase scheme.
At 48 months after therapy initiation, under prednisolone 2.5mg id and hydroxychloroquine 400mg id, clinically well, without lower limb edema; normalization of renal function; progressive decrease of proteinuria, currently 0.42gr/24h; inactive urinary sediment. Undetectable complement, ANA and anti-dsDNA. No complications related to immunosuppressive therapy.
DISCUSIÓN
La disfunción renal progresiva en el LES se caracteriza por grados variables de proteinuria, microhematuria, cilindros eritrocitarios en el sedimento urinario y hipertensión. 5,8
La NLM se presenta típicamente con proteinuria (mayoritariamente en el intervalo nefrótico). La creatinina sérica es a veces normal o ligeramente elevada. La hematuria microscópica es común. El síndrome nefrótico puede ocurrir. 6
Ausencia de hematuria significativa y elementos celulares a desfavorecer nefritis lúpica proliferativa concomitante. 8
Debe sospechosarse de NLM frente a proteinuria o síndrome nefrótico, sobre todo en las mujeres jóvenes o si LES conocido. 3 En el caso descrito se trata de un paciente del sexo masculino, en la 7 ª década de vida.
El diagnóstico fue confirmado por biopsia renal. Histológicamente caracterizado por depósitos inmunes subepiteliales y / o intramembranosos difusos, espesamiento difuso de la pared capilar glomerular y, a menudo, expansión mesangial. 4,9
En el caso descrito la inmunofluorescencia fue fundamental para el diagnóstico. 4
En comparación con NMI, la NLM presenta mayor riesgo de cronicidad, de disfunción renal y de complicaciones del síndrome nefrótico. 5,6
Antihipertensores, medidas antiproteinúricas e hipolipemiantes se recomiendan en todos los pacientes. Diuréticos con un papel importante en el control del edema. Salvaguardamos la necesidad de anticoagulación en la prevención del tromboembolismo ante proteinuria maciza o hipoalbuminemia grave. 10,11
A pesar de la inexistencia de manifestaciones extrarrenales y de laboratorio (complemento normal, anti dsDNA negativo - altamente específicos) del LES después de 48 meses, el riesgo de desarrollo de las mismas sigue presente y carece de vigilancia. 12
No existen estudios aleatorizados comparando la eficacia de terapia inmunosupresora aislada con su combinación con no inmunosupresora. 7,13
En la NLM con factores de mal pronóstico (síndrome nefrótico o agravamiento de la creatinina sérica basal o persistencia de proteinuria> 3.5g/día bajo terapia no inmunosupresora), los autores recomiendan asociación de inmunosupresión. Por otro lado, existen defensores de que en ausencia de al menos uno de los factores de mal pronóstico previos, la terapia inmunosupresora no debe utilizarse, dada su toxicidad.
Otros favorecen la inmunosupresión independientemente de la presencia de factores de mal pronóstico, dado que a diferencia de la NMI, la remisión espontánea en la NLM no ocurre. 10,14
La combinación de micofenolato de mofetilo (MMF) con glucocorticoides como terapia inicial es la recomendada. En caso de contraindicación o intolerancia a MMF, la ciclofosfamida intravenosa o un inhibidor de la calcineurina en combinación con glucocorticoides, son alternativas. 15,16
Después de iniciada la inmunosupresión los pacientes deben ser reevaluados cada 1-2 meses, ampliándose ese período si estabilidad clínica. La respuesta renal es evaluada por la creatinina sérica, la excreción urinaria de proteínas (ratio de proteína/creatinina urinaria) y el sedimento urinario. 16,17
En el caso descrito el tratamiento instituido demostró buenos resultados clínicos y de laboratorio con normalización de la función renal en 5 meses y remisión de la proteinuria.
Los autores destacan que el abordaje del paciente con NL clase V sigue siendo un desafío, desde el momento del diagnóstico hasta el reconocimiento de la evolución clínica e institución terapéutica. A pesar de la investigación exhaustiva sólo por la biopsia renal se alcanzó el diagnóstico. La edad e inmunología sérica negativa han sido probablemente dificultadores de la investigación diagnóstica.
DISCUSSION
Progressive renal dysfunction in SLE is characterized by varying degrees of proteinuria, microhematuria, erythrocyte cylinders in the urine sediment and hypertension. 5,8
NLM typically presents with proteinuria (mostly in the nephrotic range). Serum creatinine is sometimes normal or slightly elevated. Microscopic hematuria is common. Nephrotic syndrome may occur. 6
Lack of significant hematuria and cellular elements to disadvantage concomitant lupus proliferative nephritis.8
Proteinuria or nephrotic syndrome should be suspected, especially in young females or if known SLE. 3 In the case described it is a male patient, in the 6th decade of life.
The diagnosis was confirmed by renal biopsy. Histologically characterized by diffuse subepithelial and/or intramembranous immune deposits, diffuse thickening of the glomerular capillary wall and, often, mesangial expansion. 4,9
In the described case, immunofluorescence was essential for diagnosis.4
Compared with IMN, MLN presents an increased risk of chronicity, renal dysfunction and nephrotic syndrome complications. 5,6
Antihypertensives, antiproteinuric and lipid lowering measures are recommended in all patients. Diuretics with important role in the control of edema. We safeguard the need for anticoagulation in the prevention of thromboembolism in the presence of massive proteinuria or severe hypoalbuminemia.10,11
Despite the absence of extrarenal and laboratory manifestations (normal complement, anti-dsDNA negative - highly specific) of SLE after 48 months, the risk of developing them is still present and requires monitoring. 12
There are no randomized trials comparing the efficacy of immunosuppressive therapy alone with its combination with nonimmunosuppressive. 7,13
In MLN with poor prognostic factors (nephrotic syndrome or worsening of baseline serum creatinine or persistence of proteinuria> 3.5 g/day under nonimmunosuppressive therapy), authors recommend association of immunosuppression.
On the other hand, there are proponents that in the absence of at least one of the previous poor prognostic factors, immunosuppressive therapy should not be used because of its toxicity. Others favor immunosuppression regardless of the presence of poor prognostic factors, since unlike IMN, spontaneous remission in NLM does not occur. 10,14
The combination of mycophenolate mofetil (MMF) with glucocorticoids as initial therapy is the recommended one. If MMF contraindication or intolerance, intravenous cyclophosphamide or a calcineurin inhibitor in combination with glucocorticoids are an alternative.15,16
After initiation of immunosuppression, patients should be re-evaluated every 1-2 months, and this period should be extended if clinical stability. Renal response is assessed by serum creatinine, urinary protein excretion (urinary protein/creatinine ratio), and urinary sediment. 16,17
In the described case, the established therapy demonstrated good clinical and laboratory results with renal function normalization at 5 months and proteinuria remission.
The authors emphasize that the approach of the patient with Lupic Nephritis class V remains a challenge, from the moment of diagnosis until the clinical evolution recognition and therapeutic institution. Despite the exhaustive investigation, the diagnosis was only achieved by renal biopsy. Age and negative serum immunology probably added as difficulties in the diagnostic investigation.
Declaracion de conflicto de interés
Ninguno declarado.
Conflict of interest statement
None declared.
Edición Marzo 2019
Figura 1
Organización genómica de seis genes de colágeno tipo IV que están distribuidos en los cromosomas 13, 2 y X. Los 5 exones en la porción 3' terminal de COL4A5, codifican el dominio NC1 de α5 (IV) (Kashtan y Michael, 1996).
Recuperado de "Revisión sobre la pérdida auditiva en el síndrome de Alport, analizando los aspectos clínicos, genéticos y biomoleculares", de Alves, F (Nov./Dec. 2005).
Figura 2 - IFx400: Depósitos granulares y gruesos de C3, C4, C1q, IgA, IgG e IgM
Figure 2 - IFx400: Granular and coarse deposits of C3, C4, C1q, IgA, IgG and IgM
Tabla 1. Distribución de pacientes participantes por provincias
Tabla 2: Fórmulas utilizadas para estimar el Filtrado Glomerular.
Tabla 3: Distribución del índice de masa corporal (IMC) en los participantes.
Tabla 4: Distribución del índice de masa corporal según sexo de los pacientes
Tabla 5: Prevalencia de factores de riesgo asociados a la participación de los pacientes, según sexo.
Tabla 6: Distribución de los resultados de RAC según rangos de medición.
Tabla 7: Porcentaje de pacientes estudiados según rango etario
Tabla 8: Resultados de RAC según edad de los pacientes
Tabla 9: Relación de pacientes con resultados de RAC alterado según sexo y edad
Tabla 10: Distribución de los resultados del eFG según ambas ecuaciones
Tabla 11: Comparación de medias de ambas fórmulas: MDRD-4 y CKD-EPI
Tabla 12: Comparación de diferencias emparejadas entre ambas fórmulas: MDRD-4 y CKD-EPI
Tabla 13: Cuadro de severidad de riesgos utilizando la fórmula MDRD-4, en frecuencias y porcentajes.
Tabla 14: Cuadro de severidad de riesgos utilizando la fórmula CKD-EPI, en frecuencias y porcentajes.
Tabla 15: Comparación de riesgos según fórmulas MDRD-4 y CKD-EPI.
Tabla 16: Distribución según provincias de los pacientes con RAC alterado.
Cuadro 2
Gráfico 1: Concordancia entre ambas fórmulas: MDRD-4 y CKD-EPI
Clasificación de riesgos según KDIGO 2012
Bibliografía.
1. Urrego-Díaz, J, Landinez-Millán G & Lozano-Triana C. Síndrome de Alport: reporte de caso y revisión. Rev Fac Med. 2015; 63(1):143-149. doi:https://doi.org/10.15446/revfacmed.v63n1.45007
2. Savige J, Gregory M, Gross O, Kashtan C, Ding J, Flinter F. Expert guidelines for the management of Alport Syndrome and thin basement membrane nephropathy. J Am Soc Nephrol. 2013; 24:364–375.
3. Medeiros-Domingo M, Fuentes Y, García-Roca P, Hernández AM, Morán- Barroso VF, Velásquez-Jones. Bol Med Hosp Infant Mex. 2008; 65(5):231- 240.
4. Rubio, R. Estudio clínico del sindrome de alport autosómico recesivo. (s. E. Nefrología, ed.) Nefrología. 2013;33(2).
5. Valle AR, Gallart MDM, Martín MTC, Roldan MLJ. Sindrome de Alport ligado al cromosoma X. Laboratorio y enfermedad. Casos clínicos. 2010, p. 116.
6. Torra R, Ars E, Tazón B. El síndrome de Alport. Nefrología 2003; 23 (S1):29-39
7. Kashtan CE, Ding J, Garosi G, Heidet L, Massella L, Nakanishi K, et al. Alport syndrome: a unified classification of genetic disorders of collagen IV
α345: a position paper of the Alport Syndrome Classification Working Group. Kidney Int. 2018; 93(5):1045-1051.
8. Gross O, Kashtan CE, Rheault MN, Flinter F, Savige J, Miner JH, et al. Advances and unmet needs in genetics, basic and clinical science in Alport syndrome. Report from the 2015 International Workshop on Alport syndrome. Nephrol Dial Transplant. 2016; 0:1-9. doi: 10.1093/ndt/gfw095
9. Watson S, Bush JS. Alport Syndrome. [Updated 2017 Dec 1]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2018 Jan-. Disponible en: https://www.ncbi.nlm.nih.gov/books/NBK470419/
10. Soler Pujol, G. Iotti R, Davalos D, Neil Turner A, Vilches R. Enfermedad antimembrana basal glomerular en un paciente trasplantado con enfermedad de Alport. Medicina (Buenos Aires) 1999;59:466-468.
11. Alves, F (Nov./Dec. 2005). Revision about hearing loss in the Alport’ s syndrome, analyzing the clinical, genetic and bio-molecular aspects[Grafico].Recuperado de:
http://www.scielo.br/scielo.php?pid=S0034- 72992005000600020&script=sci_arttext&tlng=en
Bibliografía.
1. Sethi S and Fervenza FC. Membranoproliferative Glomerulonephritis - A New look at an Old Entity. N Engl J Med. 2012; 366: 1119-1131. DOI: http://dx.doi.org/10.1056/NEJMra1108178
2. Sethi S. Etiology-Based Diagnostic Approach to Proliferative Glomerulonephritis. Am J Kidney Dis. 2014; 63(4): 561-566. DOI: http://dx.doi.org/10.1053/j.ajkd.2013.11.019
3. Little MA, Dupont P, Campbell E, Dorman A and Walshe JJ. Severity of primary MPGN, rather than MPGN type, determines renal survival and post-transplantation recurrence risk. Kidney Int. 2006; 69: 504-511. DOI: http://dx.doi.org/10.1038/sj.ki.5000084
4. Schena FP and Alpers CE. Membranoproliferative glomerulonephritis, dense deposit disease, and cryoglobulinemic glomerulonephritis. In: Jurgen Floege, Richard J. Johnson, John Feehally, editors. Comprehensive clinical nephrology. 3rd ed. Philadelphia, PA: MOSBY; 2010. p. 260-69
5. Mansani N, Jhaveri KD and Fishbane S. Update on Membranoproliferative GN. Clin J Am Soc Nephrol. 2014; 9: 600-608.
6. Fervenza FC, Sethi S and Glassock RJ. Idiopathic membranoproliferativa glomerulonephritis: does it exist? Nephrol Dial Transplant. 2012; 27: 4288-4294. DOI: http://dx.doi.org/10.1093/ndt/gfs288
7. Alpers CE, Smith KD. Cryoglobulinemia and renal disease. Curr Opin Nephrol Hypertens. 2008;17:243–249. doi: 10.1097/MNH.0b013e3282f8afe2
https://www.ncbi.nlm.nih.gov/pubmed/18408474
8. ‘Alchi B., Jayne D. 2010. Membranoproliferative glomerulonephritis. Pediatr. Nephrol. 25: 1409–1418. doi: 10.1007/s00467-009-1322-7
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2887509/
https://www.ncbi.nlm.nih.gov/pubmed/19908070
9. Orth SR, Ritz E. A síndrome nefrótica. N Engl J Med. 1998; 338 : 1202-1211. doi: 10.1056 / NEJM199804233381707. https://www.ncbi.nlm.nih.gov/pubmed/9554862
10. Dias, B. C., Testagrossa, L., Jorge, L., Malheiros and D., Woronik, V. 2017. Clinical and histological features of patients with membranoproliferative glomerulonephritis classified by immunofluorescence findings. Brazilian Journal of Nephrology, Vol. 39. http://dx.doi.org/10.5935/0101-2800.20170078
http://www.scielo.br/scielo.php?pid=S0101-28002017000400447&script=sci_arttext&tlng=pt
11. Cameron JS, Turner DR, Heaton J, Williams DG, Ogg CS, Chantler C, Haycock GB, Hicks J. Idiopathic mesangiocapillary glomerulonephritis. Comparison of types I and II in children and adults and long-term prognosis. Am J Med. 1983;74:175–192. doi: 10.1016/0002-9343(83)90606-X. https://www.ncbi.nlm.nih.gov/pubmed/6337487
https://dx.doi.org/10.1016%2F0002-9343(83)90606-X
12. Varade WS, Forristal J, West CD. Patterns of complement activation in idiopathic membranoproliferative glomerulonephritis, types I, II, and III. Am J Kidney Dis. 1990;16:196–206. https://www.ncbi.nlm.nih.gov/pubmed/2205097
13. Schwertz R, Rother U, Anders D, Gretz N, Scharer K, Kirschfink M: Complement analysis in children with idiopathic membranoproliferative glomerulonephritis: A long-term follow-up. Pediatr Allergy Immunol 12 : 166 –172, 2001
https://jasn.asnjournals.org/lookup/external-ref?access_num=11473682&link_type=MED&atom=%2Fjnephrol%2F16%2F5%2F1392.atom
https://jasn.asnjournals.org/lookup/external-ref?access_num=10.1034/j.1399-3038.2001.012003166.x&link_type=DOI
14. Levin A. Management of membranoproliferative glomerulonephritis: evidence-based recommendations. Kidney Int. 1999;70:S41–S46. doi: 10.1046/j.1523-1755.1999.07006.x. https://www.ncbi.nlm.nih.gov/pubmed/10369194
https://dx.doi.org/10.1046%2Fj.1523-1755.1999.07006.x
15. De S, Al-Nabhani D, Thorner P, Cattran D, Piscione TD, Licht C. Remission of resistant MPGN type I with mycophenolate mofetil and steroids. Pediatr Nephrol. 2009;24:597–600. doi: 10.1007/s00467-008-1023-7.
https://www.ncbi.nlm.nih.gov/pubmed/18972137
https://dx.doi.org/10.1007%2Fs00467-008-1023-7
16. Jones G, Juszczak M, Kingdon E, Harber M, Sweny P, Burns A. Treatment of idiopathic membranoproliferative glomerulonephritis with mycophenolate mofetil and steroids. Nephrol Dial Transplant. 2004;19:3160–3164. doi: 10.1093/ndt/gfh526.
https://www.ncbi.nlm.nih.gov/pubmed/15479745
https://dx.doi.org/10.1093%2Fndt%2Fgfh526
17. Tarshish P, Bernstein J, Tobin JN, Edelmann CM., Jr Treatment of mesangiocapillary glomerulonephritis with alternate-day prednisone—a report of the International Study of Kidney Disease in Children. Pediatr Nephrol. 1992;6:123–130. doi: 10.1007/BF00866289. https://www.ncbi.nlm.nih.gov/pubmed/1571205
https://dx.doi.org/10.1007%2FBF00866289
18. Somers M, Kertesz S, Rosen S, Herrin J, Colvin R, Palacios de Carreta N, Kim M. Non-nephrotic children with membranoproliferative glomerulonephritis: are steroids indicated? Pediatr Nephrol. 1995;9:140–144. doi: 10.1007/BF00860727.
https://www.ncbi.nlm.nih.gov/pubmed/7794705
https://dx.doi.org/10.1007%2FBF00860727
19. Faedda R, Satta A, Tanda F, Pirisi M, Bartoli E. Immunosuppressive treatment of membranoproliferative glomerulonephritis. Nephron. 1994;67:59–65. doi: 10.1159/000187889. https://www.ncbi.nlm.nih.gov/pubmed/8052369
https://dx.doi.org/10.1159%2F000187889
20. Donadio JV, Jr, Anderson CF, Mitchell JC, 3rd, Holley KE, Ilstrup DM, Fuster V, Chesebro JH. Membranoproliferative glomerulonephritis. A prospective clinical trial of platelet-inhibitor therapy. N Engl J Med. 1984;310:1421–1426.
https://www.ncbi.nlm.nih.gov/pubmed/6371535
21. Zäuner I, Böhler J, Braun N, Grupp C, Heering P, Schollmeyer P. Effect of aspirin and dipyridamole on proteinuria in idiopathic membranoproliferative glomerulonephritis: a multicentre prospective clinical trial. Collaborative Glomerulonephritis Therapy Study Group (CGTS) Nephrol Dial Transplant. 1994;9:619–622.
https://www.ncbi.nlm.nih.gov/pubmed/7970086
Bibliografía.
1 Mac-Moune Lai F, Li KM, Tang NLS, Li PKT, Lui SF, Lai KN. IgA nephropathy: a rare lesion in systemic lupus erythematosus. Mod Pathology 1995;8:5 -10
2 Churg J, Bernstein J, Glassock R. Renal Disease: Classification and Atlas of Glomerular Diseases, 2nd ed. New York, Igaku -Shoin, 1995, p. 151 -155
3 Wuning JJ, D’Agati VD, Appel GB, et al. The classification of glomerulonephritis in systemic lupus nephritis revisited. Kidney Int 2004;65:521 -530
4 Appel GB, Radhakrishnan, D’Agati V. Secondary glomerular disease. In Brenner B (ed): The Kidney. Philadelphia, Elsevier, 2004
5 Feehally J, Floege J. IgA Nephropathy and Henoch -Schonlein Nephritis. In: Feehally J, Floege J, Johnson R, editors. Comprehensive Clinical Nephrology. 3rd ed. Philadelphia, Mosby, 2007, p. 253 -264
6 Mac -Moune lai F, Lai Kn, Lee JLK, et al. Hepatitis B virus related glomerulopathy in patients with systemic lupus erythematosus. Am J Clin Pathol 1987;88:412-420
7 D’Agati V, Seigle R. Coexistence of AIDs and lupus nephritis: a case report. Am J Nephrol 1990;10:243 -247
8 Basile C, Semeraro A, Montanaro A, et al. IgA nephropathy in a patient with systemiclupus erythematosus. Nephrol Dial Transplant 1998;13:1891 -2
9 A Corrado, L Quarta, AM Di Palma, L Gesualdo, FP Cantatore. IgA nephropathy in systemic lupus erythematosus. Clinical and Experimental Rheumatol 2007;25:467 -469
10 Lipsky PE. Systemic lupus erithematosus. An autoimmune disease of B cell hyperactivity. Nat Immunol 2001;2:764 -766
11 Geddes CC, Rarta V, Gronhagen -Riska C, et al. A tricontinental review of IgA nephropathy. Nephrol Dial Transplant 2003;18:1541
12 Goshen E, Livne A, Nagy J, Sarov I, Shoenfeld Y. Antinuclear autoantibodies in sera of patients with IgA nephropathy. Nephron 1990; 55:33 -36
13 Wasicek CA, Reichlin M. Clinical and serological differences between systemic lupus erythematosus patients with antibodies to Ro versus patients with antibodies to Ro and La. J Clin Invest 1982;69:835
14 Reichlin M. Clinical significance of anti -Ro/SSA and anti -La/SSB antibodies. In: URL. http://www.uptodate.com (accessed on 08.02.2009)
15 Queffeulou G, Berentbaum F, Michel C, et al. AA amyloidosis in systemic lupus erythematosus. An unusual complication. Nephrol Dial Transplant 1998; 13:1846-1848
Bibliografía.
1- Alemano G, Celia E, Cusumano AM, Depine S, Greloni G, Inserra F, et al: Guía de práctica clínica sobre prevención y detección precoz de la enfermedad renal crónica en adultos en el primer nivel de atención. Programa nacional de garantía de calidad de la atención médica. Dirección de calidad de los servicios de salud. Ministerio de Salud de la Nación, Argentina. Marzo 2010.
2- Kidney Disease: Improving Global Outcomes (KDIGO) CKD Work Group. KDIGO 2012 Clinical Practice Guideline for the Evaluation and Management of Chronic Kidney Disease. Kidney inter., Suppl. 2013; 3:1–150.
3- Levey AS, de Jong PE, Coresh J, Gansevoort RT, Kasiske BL, Eckardt KU, et al. The definition, classification, and prognosis of chronic kidney disease: a KDIGO Controversies Conference report. Kidney Int 2011; 80: 17-28.
4- Go AS, Chertow GM, Fan D, Mc Culloch CE, Hsu CY: Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N Engl J Med 2004 Sep 23; 351(13):1296-305.
5- Cusumano AM. Enfermedad renal crónica: Necesidad de implementar programas para su detección precoz y prevención de su progresión. Acta Científica Estudiantil 2007; 5(4): 139-146.
6- Canal C, Pellicer R, Facundo C, Gracia-García S, Montañés-Bermúdez R, Ruiz-García C, et al. Tablas de estimación del filtrado glomerular mediante la nueva ecuación CKD-EPI a partir de la concentración de creatinina sérica. Nefrología 2014; 34 (2):223-9.
7- Rosa-Diez G, Varela F, Crucelegui S, Algranati S, Greloni G. Comparación entre las ecuaciones CKD-EPI y MDRD-4 para la estimación del filtrado glomerular en pacientes con enfermedad renal crónica. Medicina (Buenos Aires) 2011; 71: 323-330.
8- Stevens L, Cooper S, Singh S, Levin A. Detection of chronic kidney disease in non-nephrology practices: An important focus for intervention. BCMJ 2005; 47(6): 305-311.
9- Alcázar R, Albalate M. Nuevas fórmulas para estimar el filtrado glomerular. Hacia una mayor precisión en el diagnóstico de la enfermedad renal crónica. Servicio de Nefrología. Hospital Infanta Leonor. Madrid. Nefrología 2010; 30(2):143-6
10- Gracia S, et al. Documento de consenso: Recomendaciones sobre la utilización de ecuaciones para la estimación de filtrado glomerular en adultos. Nefrología. 2006; vol 26 (6): 658-665.
11- Rodrigo Tagle V. Treatment of hypertension in cronic kidney disease. Rev Med Clin Condes 2010; 21 (4): 541-552.
12- Morales J. Drogas Nefrotóxicas. Rev. Med. Clin. Condes 2010; 21 (4) 623-628.
13- Qaseem A, Hopkins RH Jr, Sweet DE, Starkey M, Shekelle P. Screening, monitoring and treatment of stage 1 to 3 chronic kidney disease: A clinical practice guidelines from the American College of Physicians. Ann Intern Med. 2013 Dec 17; 159 (12): 835-47.
14- Organización Mundial de la Salud (OMS) www.who.int/es/news-room/fact-sheets/detail/obesity-and-overweight - 84k (fecha de acceso: 25 enero 2016).
15- Bertakis KD, Azari R, Helms LJ, Callahan EJ, Robbins JA. Gender differences in the utilization of health care services. J Fam Pract 2000; 49 (2): 147-152.
16- Yamagata K, Ishida K, Sairenchi T, Takahashi H, Ohba S, Shiigai T, et al. Risk factors for chronic kidney disease in a community-based population: a 10-year follow-up study. Kidney Int 2007 Jan; 71(2): 159-166.
17- Atkins RC. The changing patterns of chronic kidney disease: the need to develop strategies for prevention relevant to different regions and countries. Kidney Int Suppl. 2005 Sep; (98): S83-S85.
18- Martínez ME, Plazas M, Barajas GP, Bravo AM, González C, Rodríguez A, et al. Factores de riesgo para enfermedad renal crónica en pacientes que asisten a consulta de medicina interna. Acta Méd Colombia. 2013; 38 (4): 228-232.
19- Hill N, Fatoba S, Oke J, Hirst J, O´Callaghan C, Lasserson, D. Global Prevalence of Chronic Kidney Disease- A Systematic Review and Meta-Analysis. 2016; PLoS ONE 11(7): e0158765.
20- Coresh J, Selvin E, Stevens LA, Manzi J, Van Lente F, Levey AS, et al. Prevalence of chronic kidney disease in the United States. JAMA 2007, Nov ; 298(17) 2038-47.
21- Levey AS, Lesley A, Stevens MS, Zhang YL, Castro AF, Feldman HI, et al. A new equation to estimate glomerular filtration rate. Ann Intern Med 2009; 150: 604-612.
Bibliografía.
1 - Bhinder S, Singh A, Majithia V. Membranous (class V) renal disease in systemic lupus erythematosus may be more common than previously reported: results of a 6-year retrospective analysis. Am J Med Sci 2010; 339:230.
2 - Cameron JS. Lupus nephritis. J Am Soc Nephrol 1999; 10:413.
3 - Adu D, Williams DG, Taube D, et al. Late onset systemic lupus erythematosus and lupus-like disease in patients with apparent idiopathic glomerulonephritis. Q J Med 1983; 52:471.
4 - Jennette JC, Iskandar SS, Dalldorf FG. Pathologic differentiation between lupus and nonlupus membranous glomerulopathy. Kidney Int 1983; 24:377.
5 - Donadio JV Jr, Burgess JH, Holley KE. Membranous lupus nephropathy: a clinicopathologic study. Medicine (Baltimore) 1977; 56:527.
6 - Sloan RP, Schwartz MM, Korbet SM, Borok RZ. Long-term outcome in systemic lupus erythematosus membranous glomerulonephritis. Lupus Nephritis Collaborative Study Group. J Am Soc Nephrol 1996; 7:299.
7 - Austin HA 3rd, Illei GG, Braun MJ, Balow JE. Randomized, controlled trial of prednisone, cyclophosphamide, and cyclosporine in lupus membranous nephropathy. J Am Soc Nephrol 2009; 20:901.
8 - Mercadal L, Montcel ST, Nochy D, et al. Factors affecting outcome and prognosis in membranous lupus nephropathy. Nephrol Dial Transplant 2002; 17:1771.
9 - Schwartz MM, Kawala K, Roberts JL, et al. Clinical and pathological features of membranous glomerulonephritis of systemic lupus erythematosus. Am J Nephrol 1984; 4:301.
10 - Sun HO, Hu WX, Xie HL, et al. Long-term outcome of Chinese patients with membranous lupus nephropathy. Lupus 2008; 17:56.
11 - Beck LH Jr, Salant DJ. Treatment of membranous lupus nephritis: where are we now? J Am Soc Nephrol 2009; 20:690.
12 - Pasquali S, Banfi G, Zucchelli A, et al. Lupus membranous nephropathy: long-term outcome. Clin Nephrol 1993; 39:175.
13 - Waldman M, Appel GB. Update on the treatment of lupus nephritis. Kidney Int 2006; 70:1403.
14 - Karim MY, Pisoni CN, Ferro L, et al. Reduction of proteinuria with mycophenolate mofetil in predominantly membranous lupus nephropathy. Rheumatology (Oxford) 2005; 44:1317.
15 - Moroni G, Maccario M, Banfi G, et al. Treatment of membranous lupus nephritis. Am J Kidney Dis 1998; 31:681.
16 - Appel GB, Contreras G, Dooley MA, et al. Mycophenolate mofetil versus cyclophosphamide for induction treatment of lupus nephritis. J Am Soc Nephrol 2009; 20:1103.
17 - Touma Z, Urowitz MB, Ibañez D, Gladman DD. Time to recovery from proteinuria in patients with lupus nephritis receiving standard treatment. J Rheumatol 2014; 41:688.
 Figura 1
Figura 1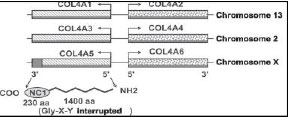
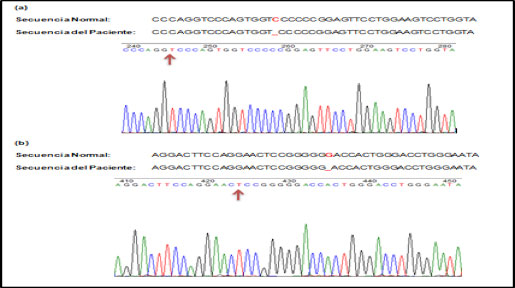
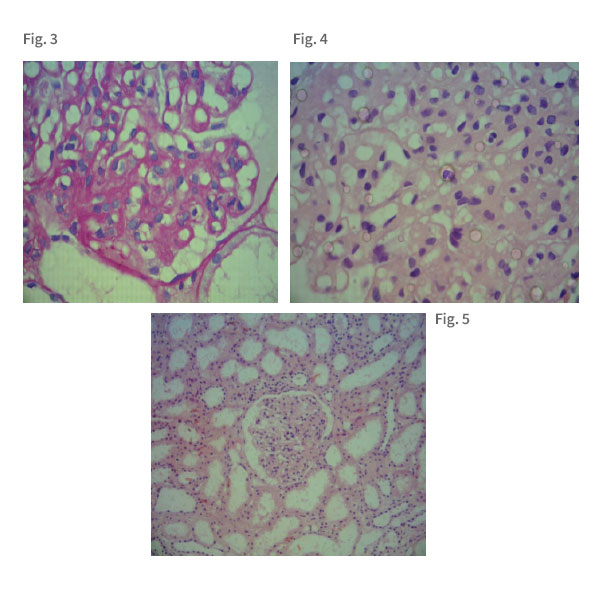
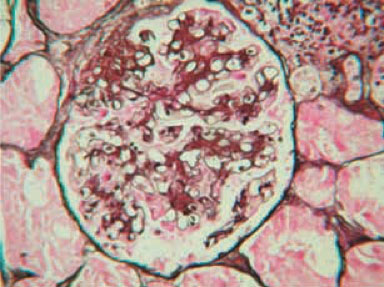
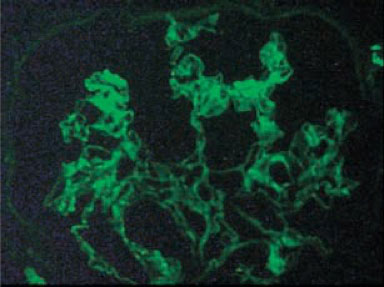
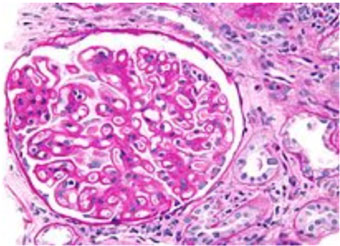
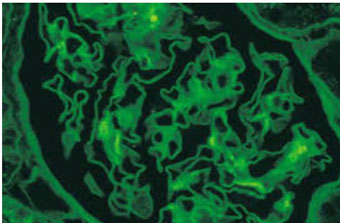
 Tabla 1. Distribución de pacientes participantes por provincias
Tabla 1. Distribución de pacientes participantes por provincias